| IQ, OQ, & PQ Validation Protocols | 您所在的位置:网站首页 › IQOQPQ › IQ, OQ, & PQ Validation Protocols |
IQ, OQ, & PQ Validation Protocols
|
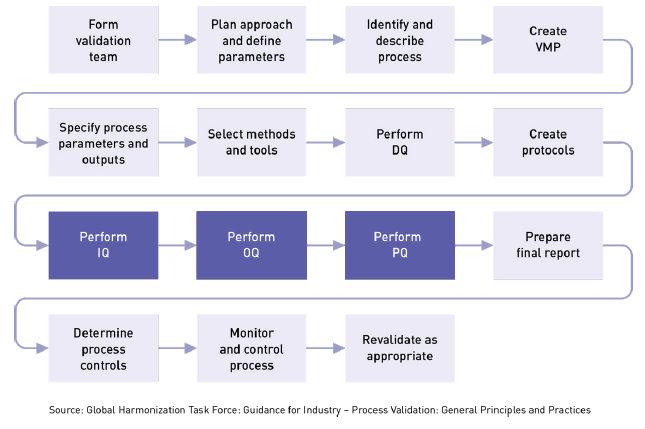
The goal is process validation is to produce a stable medical device manufacturing process that offers consistent performance. Variation is minimal and predictable. Your Process Validation Plan (PVP) will provide the framework for executing three important phases in the validation process. Installation Qualification (IQ): Is everything installed correctly? Operational Qualification (OQ): Is everything operating correctly? Are operating limits understood? Performance Qualification (PQ): Did it produce the correct result? Is the process stable and capable? If you want to learn more about process validation, check out our Process Validation Training for Medical Devices: Principles and Protocols class. What is Installation Qualification (IQ)? IQ can be executed by the facilities, engineering, production or operations groups and the basic idea is to ensure that everything is installed properly. During this qualification phase the installation of equipment, piping, services, and instrumentation will be checked against engineering drawings, piping and instrument diagrams (P&ID), and functional specifications developed during the project planning stage. During the project planning stage, all system elements, service conduits, and gauges are identified, and a documented record is prepared showing that all installed equipment satisfies your requirements. At various stages during IQ, you will need to review, check, report, and authorize protocols, documentation, procedures, equipment, specifications, and acceptance criteria for test results.
IQ touches on many tangible and intangible aspects related to installation including: Components and equipment. Software and Standard Operating Procedures (SOP). Calibration and testing. Installation. Software and systems testing. Records. Electrical. Signals and alarms. Revalidation, facility conditions, documentation.There should be an SOP, checklist or some other documented process that defines the standard installation procedure for each type of system or deliverable being installed. This is required for any equipment used in the manufacturing process. IQ verifies and documents key aspects of an installation meet approved requirements. These requirements may come from: Design specifications. System specifications. Manufacturer’s recommendations. Developers’ recommendations. Data sheets. US FDA Requirements for IQThe US FDA outlines the key areas of IQ in 21 CFR 820.70(g): 1- Maintenance schedule: Each manufacturer shall establish and maintain schedules for the adjustment, cleaning, and other maintenance of equipment to ensure that manufacturing specifications are met. Maintenance activities, including the date and individual(s) performing the maintenance activities, shall be documented. 2 – Inspection: Each manufacturer shall conduct periodic inspections in accordance with established procedures to ensure adherence to applicable equipment maintenance schedules. The inspections, including the date and individual(s) conducting the inspections, shall be documented. 3 – Adjustment: Each manufacturer shall ensure that any inherent limitations or allowable tolerances are visibly posted on or near equipment requiring periodic adjustments or are readily available to personnel performing these adjustments. 4 – Automated processes: Each manufacturer shall validate computers or data processing systems that are used as part of production processes. Consequences of Improper Medical Device IQAside from being a regulatory imperative, Installation Qualification offers some tangible benefits to the company. For instance, IQ can reduce the risk that workers did not install equipment according to the procedure. It can also reduce the chance that a potentially serious blunder will be overlooked. An example includes having no backup for components damaged or destroyed during installation, purchasing/coding software that won’t work with installed equipment, or installing equipment with voltage or amperage requirements outside the range of your existing electrical supply. In addition to IQ, you may perform Design Qualification. DQ proves that the design or selection of the equipment meets your requirements. It documents that that equipment considerations were included in design controls and the equipment requirements identified prior to purchasing. Think of DQ as the bridge from design to manufacturing. This is an important element in planning for product realization and resources required by ISO 13485:2016 (7.1 and 6.3). With IQ Complete, It’s Time to Conduct the Medical Device OQ ProcessAfter you have performed IQ and checked all aspects of installation, the next step is to perform Operational Qualification (OQ). This is where you challenge your parameters to make sure your process will result in a product that meets requirements. OQ is associated with equipment performance to ensure that the functions of machines, measuring devices, utilities, and manufacturing areas perform as intended throughout all anticipated operating ranges in the selected environment. This is typically accomplished by identifying important process variables and providing evidence that even if you produce devices at limits of those parameters they will still meet specs. The OQ process does the following: Identifies the critical operating parameters Lists the experiments to be conducted on the critical variables Describes the sequence of the experiments Lists the necessary measuring equipment Ensures the acceptance criteria for the product can be metIt also includes the procedures required to verify specific dynamic attributes of the new or modified process throughout its operating range, which may include worst-case conditions. Generally, you will start the OQ process according to plan and let it reach standard operating conditions. You will then monitor the operating parameters to ensure that the process start-up occurs as expected. Once the process is stable, you can send product through and test the final product. You can then adjust the operating conditions to test the limits of the key inputs. While the OQ is being conducted, you’ll want to perform several other checks to ensure they are operating with specified ranges. These include process controls, voltage and amperage levels, computer and software systems, environmental conditions (e.g., temperature, humidity, air quality, etc.) safety, alarms and interlocks, automation features, activity triggers, and timing. This is also an opportunity to check correct use of procedures by operating personnel. Lack of proper operational qualification can result in many problems. These might include a process that does not start up correctly or once stabilized, produces a product that does not meet your specifications. Items that have passed the IQ can falter in operation. The completion of a satisfactory OQ should permit a formal release of the Performance Qualification (PQ) process. That release should take the form of a written authorization from your validation team and management. The Completion of OQ Leads to Performance Qualification (PQ)With OQ successfully completed, you can move on to conduct PQ – the final stage in the validation process. By now all the bugs should have been worked out during IQ and OQ so that the PQ should (hopefully) proceed smoothly. During this phase you will generate evidence that your process will consistently produce an acceptable product under normal operating conditions over the long-term. PQ is performed on the manufacturing process as a whole. Components of the system or process are typically not tested individually. Performance Qualification should also include testing the system against its operational capacity but not exceeding it. It is important at this stage to ensure that all operational test data conform with predetermined acceptance criteria from the previous qualifications. Elements of a PQ ProtocolThe absence of process qualification can cause many problems including a process that will not stabilize, or a process that is stable but produces products that meet specifications intermittently. Thus, a robust process qualification protocol is important. Here are some elements that should be included in your PQ protocol: Description of the process in the regular work environment. Duration of the PQ (dependent on the nature of the process) Standard operating parameters and limits. Acceptance criteria for qualification. Performance measurement indicators and expected results. Test procedures for each item of measuring equipment. Test data to be gathered and the time frame and schedule for gathering data. Instructions on handling abnormal data and resolution procedure for unexpected results. Documented justification for using whatever data are to be used. Requirements for setting up data acquisition, whether it is manual or automated. Instructions on how test results are to be reported, whether manually or electronically. List of statistical tools that can be used to analyze the data. Contents of the PQ ReportUpon successful completion of the PQ, the process validation project will be complete and the new or modified process can be placed into routine production. Your Performance Qualification report should include statements on whether or not the PQ protocol was followed in its entirety and reasons for any deviations. You will also want to reference to all data gathered during the PQ, prepare a summary of conclusions drawn, state whether the expected results were achieved and any follow-up activities you plan to correct deviations. The following checklist for the PQ portion of process validation combines both the FDA QSR and GHTF guidance. Have the product characteristics been defined? Have the process parameters been set? Has the SOP been established and maintained? Does the SOP define the nominal values for the process parameters? Have the operators been trained? Has the method for monitoring been established? Is there a mechanism in place to evaluate proposed process changes to see if the process requires revalidation? Is there a mechanism in place to evaluate process deviations to see if the process requires revalidation?The completion of a satisfactory PQ should permit a formal release of the process for full production. The release should take the form of written authorizations and approvals from the process validation team and management. The validation team then prepares a final report on the entire process validation project and presents it to management. Final Step: Understanding When to RevalidateThis is the third post in a 4-part series. In our first post we covered the basics of process validation, and in subsequent posts we cover validation plans and protocols , and revalidation. Finish up this series by reading our final post which talks about when to revalidate. Download the entire series in one convenient PDF. If you would really like to boost your knowledge of the topic, consider our three-day class on medical device process validation. Our team is here to help. Call 1.800.472.6477 or contact us online › |
【本文地址】