| 单细胞数据分析没有思路?试试细胞轨迹分析~(内附代码) | 您所在的位置:网站首页 › 轨迹分析软件 › 单细胞数据分析没有思路?试试细胞轨迹分析~(内附代码) |
单细胞数据分析没有思路?试试细胞轨迹分析~(内附代码)
|
上文有提过,细胞轨迹分析目前在细胞分化、谱系发育、肿瘤/疾病微环境中免疫细胞的动态变化研究中均有广泛应用,大致可以概括为以下三种应用场景:
1、细胞分化过程重现
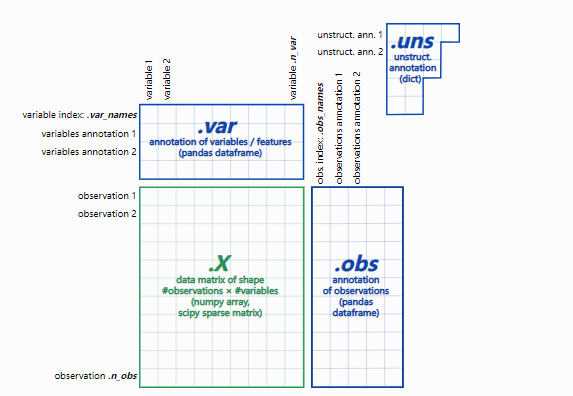
基于单细胞测序分析数据,对其他技术手段获取的已知细胞分化关系进行验证。 2020年Nature Communication发表的Single-cell RNA-seq reveals that glioblastoma recapitulates a normal neurodevelopmental hierarchy[5],利用RNA速度表征了胶质母细胞瘤的分化过程,证实了胶质母细胞瘤层次结构顶点是肿瘤祖细胞,星形细胞、间充质、少突胶质细胞和神经元癌细胞比肿瘤祖细胞的分化程度更高,并且在每个患者中均可见特定细胞分化方向(图3)。  图 3 胶质母细胞瘤RNA速度分析[5] 2020年Cell stem cell发表的The Dynamic Transcriptional Cell Atlas of Testis Development during Human Puberty[6],研究根据前期研究基础将人睾丸生殖细胞分为对应4个发育阶段的细胞亚型:未分化的精原细胞、分化的精原细胞、精母细胞、精子细胞;同时利用拟时序分析,证实了生殖细胞由未分化的精原细胞向精子细胞分化的发育轨迹(图4)。 图4 人睾丸生殖细胞亚群及分化轨迹鉴定[6] 2、潜在的细胞分化路径挖掘除了鉴定已知的细胞分化过程,通过细胞轨迹分析也能推断未被报道的、可能的未知细胞分化路径,如神经干细胞发育、肿瘤/疾病微环境中免疫细胞分化过程,甚至挖掘到一些稀少的、常规技术手段很难获得的中间状态细胞。 2021年Nature Communication发表的Single-cell sequencing of immune cells from anticitrullinated peptide antibody positive and negative rheumatoid arthritis[7],研究首先通过比较两种亚型类风湿性关节炎(RA)患者与健康对照外周血免疫细胞图谱差异,关注到了B细胞类型丰度变化,发现ACPA-RA组IGHG4+ plasma B细胞明显升高,HLA-DRB5+ plasma B细胞明显降低(图5,左图);随后对B细胞进行拟时序分析探究B细胞亚型分化关系,发现在健康对照和RA患者之间B细胞亚型存在不同的分化路径:在健康对照中naïve B细胞有两条分化路径,1条是分化为HLA-DRB5+plasma B细胞再分化为plasma B细胞,另1条分化为Mem-unsw B细胞到Mem-sw B细胞;而RA患者中,naïve B细胞直接分化为IGHG4+ plasma B细胞(图5,右图)。  图5 类风湿关节炎患者外周血B细胞分析[7] 胶质母细胞瘤 (GBM) 可以根据基因表达分为不同的亚型,但尚未鉴定出经典亚型的胶质瘤干细胞样细胞 (GSC),2019年Cancer Discovery发表的The Phenotypes of Proliferating Glioblastoma Cells Reside on a Single Axis of Variation[8],对IDH 野生型 GBM单细胞数据进行 RNA 速度分析,鉴定到了GBM的肿瘤干细胞样细胞 (GSC)群体逐渐从间充质表型mGSC 过渡到原神经表型pGSC的中间群体,在这个中间群体中,mGSC 标记(如CD44、CHI3L1)高表达;而pGSC 标记物(如DLL3、PDGFRA)的表达较低,但具有较高的正表达速度(图6)。 图6 胶质母细胞瘤干细胞样细胞分化轨迹[8] 3、细胞分化过程调控机制解析通过细胞轨迹分析,分析处于重要分化节点的关键细胞亚群,寻找随着分化时间呈现特定表达模式的关键基因,解析驱动细胞亚群分化的调控机制。 2021年 Nature发表的Single-cell transcriptomic characterization of a gastrulating human embryo[9],为了探索原肠胚形成过程中外胚层的变化,利用RNA速度分析了外胚层(Epiblast)、外胚层(羊膜/胚胎)(Ectoderm (amniotic/embryonic))、新生中胚层(Nascent mesoderm)、原条(Primitive Streak),由Epiblast起,发生两个分支,一侧由Primitive Streak发育到Nascent mesoderm,一侧发育到ectoderm(图7,左图);随后利用拟时分析推断Epiblast分化过程中的基因表达变化,检测到ectoderm marker genes(DLX5、TFAP2A 和GATA3)表达上调,而早期神经诱导marker genes(SOX1、SOX3 和PAX6)和分化的神经元marker genes(TUBB3、OLIG2和NEUROD1)表达极低甚至检测不到,这表明CS7中神经分化尚未开始(图7,右图)。  图7 RNA速度分析解析原肠胚分化轨迹[9] 如何进行RNA速度分析? 1、软件安装:pip install -U scvelo #需要3.6以上版本python pip install velocyto 2、adata 数据结构: – adata.X:数据矩阵 – adata.obs:细胞的注释 – adata.var:基因的注释 – adata.nus:非结构化注释,例如图 – adata.layers:附加层数据,如spliced/unspliced矩阵
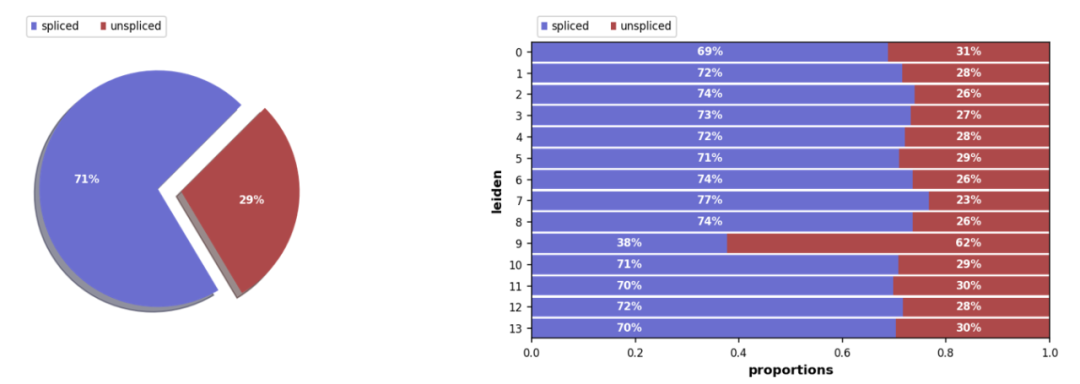
import seaborn as sns import scvelo as scv import pandas as pd import scanpy as sc import numpy as np import loompy scv.settings.verbosity = 3 # show errors(0), warnings(1), info(2), hints(3) scv.settings.presenter_view = True # set max width size for presenter view scv.set_figure_params(‘scvelo’) # for beautified visualization 4、计算获取unspliced/spliced矩阵 ## 运行run10x /software/python3.9.6/bin/velocyto run10x \ # 相应的repeatmasker的重复序列注释文件(可从USCU中获取) # http://genome.ucsc.edu/cgi-bin/hgTables -m RMSK cellranger_results # cellranger的输出结果路径 sample_name # 样本名 seurat_GTF # 用于cellranger定量的基因组注释文件 ## 输出结果,在cellranger的结果中会创建单个样本的velocyto文件夹,其中的loom文件即为最终结果 data/SC/Normal1-sc/velocyto: Normal1-sc.loom data/SC/Normal2-sc/velocyto: Normal2-sc.loom data/SC/Normal3-sc/velocyto: Normal3-sc.loom data/SC/Normal4-sc/velocyto: Normal4-sc.loom 5 、loom文件的读取及合并 # 读取单个样本loom文件 Normal_loom = sc.read(‘./Normal1.loom’, sparse=True, cache=True) # 合并同组样本的loom文件 import loompy Normal_loom_list = [Normal1-sc.loom, Normal2-sc.loom, Normal3-sc.loom, Normal4-sc.loom] loompy.combine(Normal_loom_list, output_file = ‘Normal_combined.loom’) # 读取 Normal_loom = sc.read(‘./Normal_combined.loom’, sparse=True, cache=True) 6 导入Seurat对象的降维聚类结果 使用scVelo进行分析时需要对数据进行降维聚类,若上游分析的降维聚类注释结果基于Seurat,可以直接将Seurat的降维聚类结果添加到adata对象中,下面方法仅供参考。此外,如果上游分析基于scanpy,则可以直接调用scanpy的降维聚类相关的方法。注:此处并没有进行注释,在实际操作中将 seurat_cluster替换为注释后的信息即可,导入seurat之后的所有调用的方法中的groupby参数设置为 seurat_cluster。 library(Seurat) library(tidyverse) [email protected] %>% rownames_to_column(var = ‘barcodes’) %>% write.table(‘seurat_meta_df.txt’, sep = ‘\t’, quote = F) seurat_obj@[email protected] %>% rownames_to_column(var = ‘barcodes’) %>% write.table(‘seurat_umap.txt’, sep = ‘\t’, quote = F) def using_seurat_meta_umap(loom_file, seurat_meta_df, seurat_umap): # 读入seurat的meta.data以及umap降维结果 seurat_meta_data_raw = pd.read_csv(seurat_meta_df) seurat_umap_raw = pd.read_csv(seurat_umap) # loom文件原始的index顺序 anndata_index = loom_file.obs.index.values # 合并 meta_data = loom_file.obs.reset_index(). \ merge(seurat_meta_data_raw, left_on=”CellID”, right_on=”barcodes”, how=’left’). \ set_index(‘CellID’).reindex(anndata_index) meta_data[‘seurat_clusters’] = meta_data[‘seurat_clusters’]. \ apply(lambda x: “%d” % int(x) if ~np.isnan(x) else x).astype(‘category’) loom_file.obs = meta_data loom_file = loom_file[loom_file.obs.dropna().index, :] loom_file.obsm[‘umap’] = seurat_umap_raw.set_index(‘barcodes’). \ reindex(loom_file.obs.index).to_numpy().astype(‘float64’) return loom_file Normal_loom = using_seurat_meta_umap(Normal_loom, ‘seurat_meta_df.txt’, ‘seurat_umap.txt’) # 查看是否替换成功,与seurat结果进行比较 sc.pl.umap(Normal_loom, color=’seurat_clusters’) 分析结果展示 拼接/未拼接计数的比例,通常有 10%-25% 的未拼接分子包含内含子序列。 scv.pl.proportions(Normal_loom, groupby=’seurat_clusters’)
RNA-速度:速度是基因表达空间中的向量,代表单个细胞运动的方向和速度。速度是通过模拟剪接动力学的转录动力学获得的,对于每个基因,拟合了成熟(未剪接)和成熟(剪接)mRNA 计数的稳态比率,这构成了恒定的转录状态。正速度表明基因被上调,这种情况发生在该基因未剪接 mRNA 丰度高于预期的稳定状态的细胞中。相反,负速度表明基因被下调。 # 预处理 scv.pp.filter_genes(Normal_loom, min_shared_counts=20) scv.pp.normalize_per_cell(Normal_loom) scv.pp.filter_genes_dispersion(Normal_loom, n_top_genes=2000) scv.pp.log1p(Normal_loom) scv.pp.filter_and_normalize(Normal_loom, min_shared_counts=20, n_top_genes=2000) # 运行scVelo动态模型,如果算力不够可以考虑静态或者随机方法 scv.tl.recover_dynamics(Normal_loom, n_jobs=30) scv.tl.velocity(Normal_loom, mode=’dynamical’) scv.tl.velocity_graph(Normal_loom, n_jobs=n_jobs) # 保存计算结果 Normal_loom.write(‘Normal_dy_model.h5ad’, compression=’gzip’) scv.pl.velocity_embedding_stream(Normal_loom, basis=’umap’, color=’seurat_clusters’) scv.pl.velocity_embedding(Normal_loom, arrow_length=3, arrow_size=2, show=False, color=’seurat_clusters’)
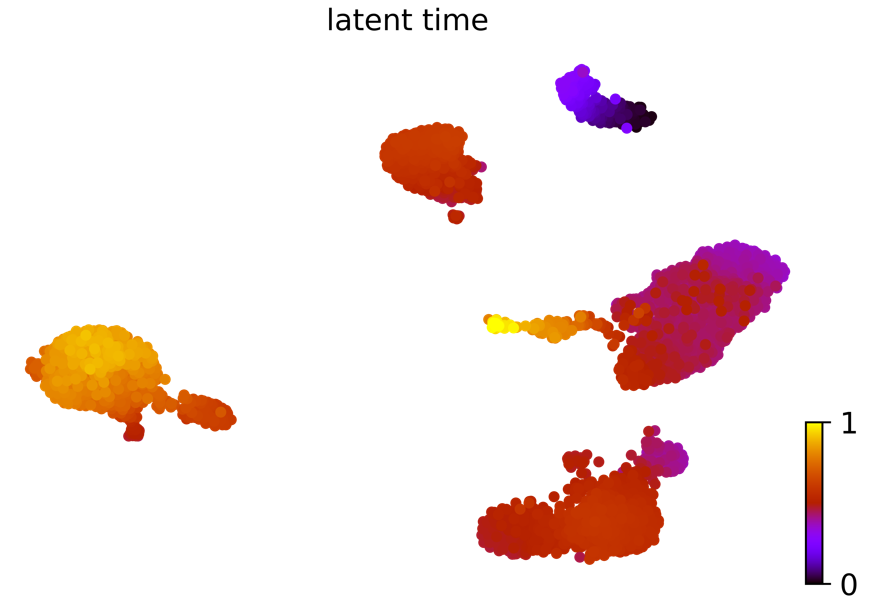
潜伏时间:动力学模型描述了细胞过程的潜伏时间(这个潜伏时间代表细胞的内部时钟,并近似于细胞在分化时经历的真实时间,仅基于其转录动力学。) scv.tl.latent_time(Normal_loom) scv.pl.scatter(Normal_loom, color=’latent_time’, color_map=’gnuplot’, size=80, show=False)
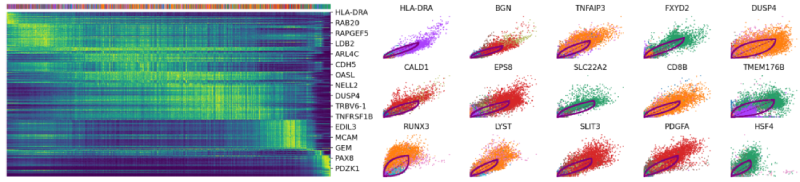
驱动基因:显示出明显的动态行为,并通过动态模型中的高可能性特征系统地检测到。 top_genes = Normal_loom.var[‘fit_likelihood’]. \ sort_values(ascending=False).index[:300] scv.pl.heatmap(Normal_loom, var_names=top_genes, sortby=’latent_time’, col_color=’seurat_clusters’, n_convolve=100) scv.pl.scatter(Normal_loom, basis=top_genes[:15], ncols=5, frameon=False, color=’seurat_clusters’)
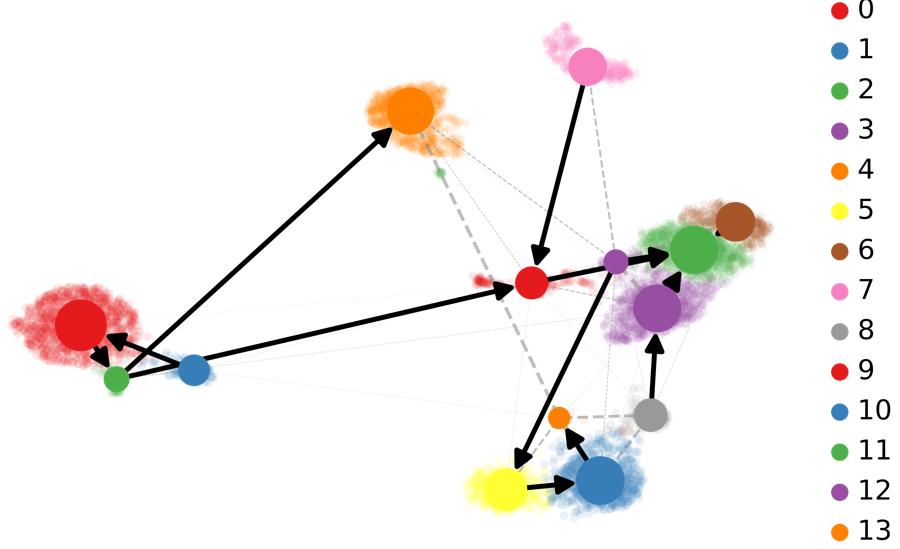
基于RNA-速度的PAGA分析:PAGA提供了轨迹推断构图的方法,其中加权边对应于两个集群之间的连接。在这里,PAGA通过速度推断的方向性进行了扩展。 scv.tl.paga(Normal_loom, groups=’seurat_clusters’) scv.pl.paga(Normal_loom, basis=’umap’, size=50, alpha=.1, min_edge_width=2, node_size_scale=1.5)
|
【本文地址】