| 科学网 | 您所在的位置:网站首页 › 质谱图相对强度是什么意思啊 › 科学网 |
科学网
|
干货篇:蛋白质谱之“搜库”
已有 24426 次阅读 2018-11-21 11:08 |个人分类:蛋白质谱|系统分类:论文交流
此文1900字,阅读需要7分钟。 什么是最常见的蛋白质谱和Shotgun? 蛋白质谱已经应用在各个研究领域,但是最常用的还是Shotgun法鉴定蛋白, 下图1中是鸟枪法蛋白质组学的实验步骤和常用的分析流程,样本蛋白经过蛋白酶水解后成肽段,经过一维或者多维的色谱法分离,肽段被电离后被碎裂形成特征串联质谱谱图,利用自动数据比对程序,将质谱谱图转变成肽段序列。然后对肽段组装和拼接进行验证,将错误的肽段信息滤除,已经被鉴定的肽段序列用来推断样本中有哪些蛋白质,当然一些肽段序列可能出现在不止一个蛋白中,这当然也会使这推断过程更加复杂。
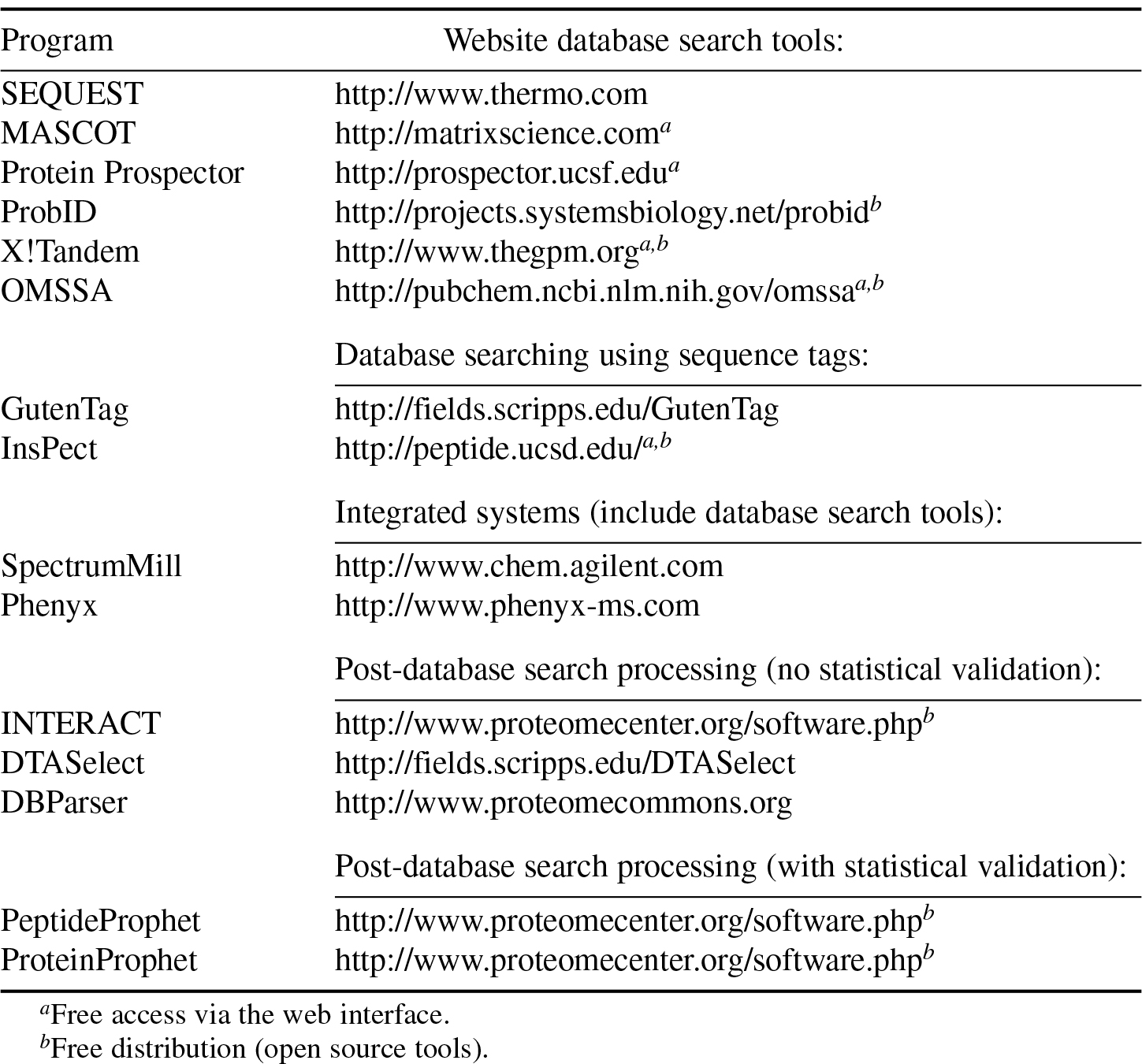
图1 搜库是个什么概念? 在一次常规的蛋白质谱实验中,搜库是比较重要的一步,质谱可以在一小时内生成成千上万张谱图,人工一个个去比对翻译这些谱图是根本不现实的。所以才会出现搜库这个概念,通过计算机软件和程序对产生的谱图进行自动化比对出检测样本中的肽段序列。那么现有计算方法可以粗略的分为三大类。 第一类:谱图直接被转换成肽段,不需要参考任何数据库,通常所说的蛋白de novo测序法; 第二类:实验谱图与数据库理论谱图比对,获取肽段序列; 第三类:是一种混合的搜库方法,采取部分目的序列片段进行比对,这是一种具有容错率较高的数据比对方法。 当然目前主流的搜库还是以第二种为主。 前面说到质谱产生这么多谱图,人工解谱无异于天方夜谭,那么这就需要搜库软件来帮助我们完成这项工作,常用的搜库软件有哪些,具体见下表。 Table:常用搜库软件
做完蛋白质谱分析后,会有很多科研工作者比较关注可信度,打分等一系列问题。打分机制是被用来比较谱图之间相似程度的一种方法,候选肽段根据计算机给出的评分进行依次排布,打分最高的肽段序列(最匹配的)将被选来做进一步分析用。 搜库参数设置 大部分数据搜索工具可以根据肽段碎裂离子类型进行设置,例如通常说的CID碎裂产生b离子和y离子,HCD碎裂还能产生其他的a, c, x, z离子。 另外一个参数是肽段质量的计算(单一同位素质量或平均质量),质谱并不直接检测肽段的质量,而是通过检测肽段的质荷比(m/z),然后根据肽段的电荷数,计算出来肽段的质量。 肽段离子带点荷数,前面提到如果要计算出肽段质量,需要知道电荷数,大多数低分辨质谱仪器只能区分出单电荷和多电荷的带电离子,但是不能轻易的判断出多电荷离子的具体电荷数,这时需要推断其带2个电荷,3个电荷的情况(因为是胰酶酶切,所以电荷数超过3的情况不常见),搜库完成后,还要选择评分最高的作为最佳匹配肽段。 母离子质量误差,根据打分选取得分最高的肽段后,质量误差则是根据使用的质谱分析器而定,如果使用的低分辩的离子阱,那么通常质量误差在2-3Da左右,TOF为基础的质谱仪,误差在0.1Da左右,当然更精准的傅里叶转换质谱,误差在0.05Da。 酶解消化的约束,许多蛋白水解酶特定识别不同的蛋白中酶切位点,例如最常见的胰酶(Trypsin),识别精氨酸(R)和赖氨酸(K),最后所有多肽的C端都是以K或者R结尾,且肽段内部不会有K或者R残基。所有的数据库搜索软件都有酶的选择作为其中一个参数,这样可以减少搜库的时间还可以限制候选肽段中出现错误的个数。 化学和翻译后修饰,大部分软件都有此修饰选项,例如:oxidation,methylation等等,一些被称为是化学修饰,例如所有蛋白的半胱氨酸上都可以发生修饰,而只有部分氨基酸上可以发生可变修饰例如甲基化修饰(Met+16Da)。 数据库怎么选? 对于一些生物体,例如人,有许多种数据可以参考:NCBI Entrez Protein database, NCBI RefSeq database和 Uniprot数据库(包括Swiss-Prot及其补充TrEMBL)。当然还有其他许多数据库都可以从网络获取,每一个数据库都不同在其条目完整性,信息丰富程度,以及肽段注释质量。至于怎么选要根据具体的实际操作经验,例如研究比较关注肽段的多态性(例,某些因基因序列发生突变导致的疾病),那么就可采用较大的数据库,例如Entrez Protein,那么也会有其对应的缺点,就是会有一些冗余的肽段序列,且EntrezProtein里面有些信息注释并不是十分完善。因此需要科研工作者进行人工分析,排除无生物作用的冗余肽段序列,这样的工作量必然是十分巨大的。当然数据库越大,那么容错率越高,越容易检测到得分高的肽段,但是并不一定是正确匹配的肽段。 对于一些数据库中没有的肽段序列,新的可变剪接体或者其他序列多态性,基因组数据库也可被用来作为蛋白数据库进行搜库,但是前提是要将DNA序列翻译成蛋白质序列,这一过程是比较复杂的,当然最后据此搜库得到的肽段信息也能是不准确的。因此,如果用蛋白数据没有发现期望的肽段序列,基因组数据搜库是最后的选择。
错误匹配的多肽序列的几种原因
前面讲过所有的谱图会根据数据库进行比对,然后确定最佳的候选肽段,但是这个结果不一定是完全准确的,其中原因有大概以下几点:
打分机制的缺陷。 有些情况下,错误的肽段序列打分有可能比正确的打分要高,这是为什么呢? 原因主要是因为打分机制被用在简单表示肽段发生碎片化的过程,特别是在理论的谱图碎片产生过程,所有的离子片段强度被认为产生了谱图并具有相同的强度。但是实际情况过程中,离子片段的强度则是依赖于氨基酸所在肽键的位置。例如,肽段N端的脯氨酸肽键断裂,会产生一个信号很强的离子片段。肽段碎裂化学通常被质谱专家们利用在人工肽段谱图确认上,因此还有很多研究需要开展将这些特殊现象整合在打分机制中。 低质量的谱图,如果谱图质量较低,那么也很难确定出正确的肽段序列,例如有很多噪音信号,也许是在样本前准备过程中引入的污染物。 多重肽段离子片段,当一些复杂的肽段混合物进行分析时,两个不同的肽段可能会有相同的质荷比,因为大部分搜库软件默认为谱图来自于单一的前体离子,因此经常会导致错误匹配的肽段。 同源肽段的原因,另外一个问题就是数据库里面的不同但是同源的肽段。这个问题在特别是在真核生物中尤其明显,比如D/N,E/Q/K这几种氨基酸组合不能通过低分辨的质谱仪区分,I/L这两种氨基酸不能通过质谱区分。那么如果肽段之间具有同源性且相同的分子质量,因此有些情况下错误匹配肽段(同源)可能打分比正确的还高。所以需要具体操作人在搜库之前考虑到此情况。 错误的肽段电荷数和质量,当带点状态是多重的,那么谱图通常需要进行两次搜库,然而如果带4个或者更多电荷数,那么软件不能区分。同样吐过质谱仪器可能还会选择一些同位素峰,例如含有一个或者多个C13的肽段离子,当这种情况发生时,从质荷比计算得出的质量就是错误的。那么即便数据库中的序列是正确的,也是不能找到正确的肽段的,除非对搜库时提高对应的容错率。 受限制的数据库搜索,因为当进行翻译后修饰以及各种化学修饰搜库时,会非常耗时耗力,因此大部分情况下都是采用不加修饰的搜库,或者搜索最常见的修饰。同样的,数据库搜索也受到酶切方式的限制。因此如果一个肽段离子带有未明确的修饰或者不同的断裂位点,那么这个肽段谱图就不会被找到。当然现在的几种搜库软件可以进行多步骤分析,例如第一步就是确定酶切位点,但是不允许添加任何修饰信息,然后再进行修饰位点检测。当然这些额外的搜索至少是在最初的搜库中发现匹配分数最高的肽段。 序列的多样性和新发现肽段,这个在最前面讲到,用基因组数据进行搜索,为了找到新的未知肽段,或者使用其他非数据库依赖的鉴定方法去进行de novo测序,这个也会造成一些错误肽段信息。 综上基本是搜库过程中可能出现的疑问和问题,后续文章还会更新相关内容,也希望感兴趣的科研工作者提出宝贵问题,进一步交流,不当之处还恳请指正。 参考文献: Nesvizhskii, A. I. and Aebersold, R. Analysis, statistical validation and dissemination of large-scale proteomics datasets generated by tandem MS. Drug Disc. Today 9, 173–181 Dancik, V., Addona, T. A., Clauser, K. R., Vath, J. E., and Pevzner, P. A. De novo peptide sequencing via tandem mass spectrometry. J. Comput. Biol. 6, 327–342. Eng, J. K., McCormack, A. L., and Yates, J. R. An approach to correlate tandem mass spectral data of peptides with amino acid sequences in a protein database. J. Am. Soc. Mass Spectrom. 5, 976–989. Zhang, N., Aebersold, R., and Schwikowski, B. ProbID: a probabilistic algorithm to identify peptides through sequence database searching using tandem mass spectral data. Proteomics 2, 406–1412 Sadygov, R. G. and Yates, J. R. A hypergeometric probability model for protein identification and validation using tandem mass spectral data and protein sequence databases. Anal. Chem. 75, 3792–3798. Frank, A., Tanner, S., Bafna, V., and Pevzner, P. Peptide sequence tags for fast database search in mass-spectrometry. J. Proteome Res. 4, 1287–1295 Apweiler, R., Bairoch, A., and Wu, C. H. Protein sequence databases. Curr. Opin. Chem. Biol. 8, 76–80. 原文链接 AIMS---蛋白质谱服务商,提供快速便捷的 LC-MS/MS 蛋白定性/定量检测。(官网建设中) https://blog.sciencenet.cn/blog-3402731-1147389.html 上一篇:去污剂对膜蛋白复合物的影响下一篇:工具篇:质谱上机前酶解处理方法之比较 收藏 IP: 183.194.173.*| 热度| |


【本文地址】