| 神经科临床病例讨论:行走缓慢2年,双下肢无力1年 | 您所在的位置:网站首页 › 胫骨前肌酸胀麻木 › 神经科临床病例讨论:行走缓慢2年,双下肢无力1年 |
神经科临床病例讨论:行走缓慢2年,双下肢无力1年
|
根据患者临床表现、家族史、神经电生理学、影像学检查,结合肌肉组织活检术和基因检测,最终明确诊断为:遗传性包涵体肌病性GNE肌病(亦称股四头肌不受累的空泡肌病或伴镶边空泡的远端肌病)。治疗方案采取以辅酶Q10 10mg/次(3次/d)、维生素B1 10mg/次(3次/d)、叶酸5mg/d、甲钴胺0.50mg/次(3次/d)、左卡尼汀1g/次(3次/d)等神经营养药物对症支持治疗。患者间断口服上述药物3个月,症状无明显变化。目前仍在随访中。
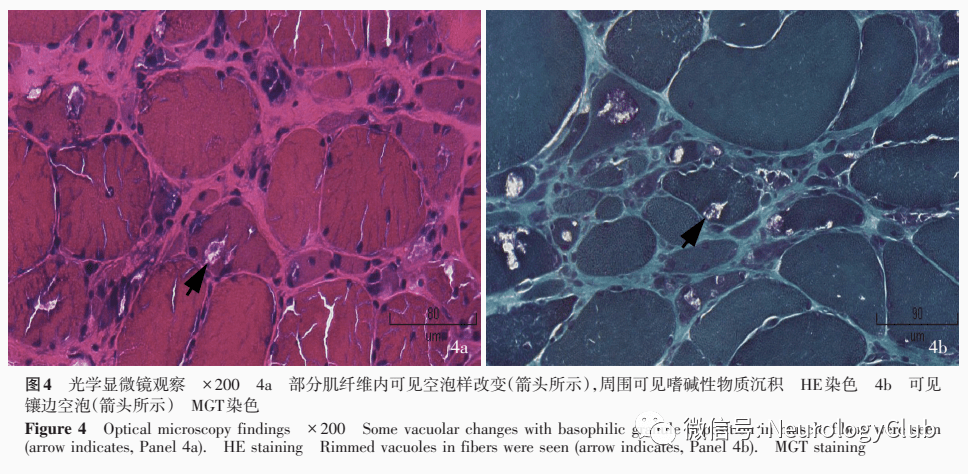
临床讨论 神经科主治医师(1)定位诊断:患者双下肢无力伴肌萎缩,肌张力降低,感觉系统检查未见异常,腱反射减弱,病理征未引出,结合肌电图、肌肉MRI检查定位于周围神经、前角细胞和肌肉,经进一步肌肉组织活检,综合定位于肌肉病变,双侧小腿前部肌群受累明显,双侧股四头肌无明显受累。 (2)定性诊断:患者为青年男性,慢性病程,呈进行性加重,临床主要表现为双下肢无力,以远端显著,双侧股四头肌不受累,伴肌萎缩,无其他系统受累表现,有家族史,血清肌酸激酶轻度升高,肌肉组织活检可见镶边空泡、病变肌肉组织无明显炎症性改变,基因检测GNE基因突变,结合组织病理学和基因检测结果,明确诊断为遗传性包涵体肌病性GNE肌病,亦称为股四头肌不受累的空泡肌病或伴镶边空泡的远端肌病。 神经科教授患者为青年男性,慢性发病,病程2年余,病情进行性加重,临床主要表现为双下肢无力和肌萎缩,尤以远端显著,双侧股四头肌不受累,血清肌酸激酶轻度升高,考虑远端型肌病,疾病分型依靠临床表现、肌肉组织活检和基因检测结果。进一步的肌肉组织活检呈现慢性肌肉病表现,可见较多镶边空泡,病变肌肉组织无明显炎症性改变,基因检测显示GNE基因c.622C>T(p.Arg208Cys)和c.649T>C(p.Tyr217His)复合杂合突变,可以确诊为遗传性包涵体肌病性GNE肌病。 该病以股四头肌不受累为特点,故又称为股四头肌不受累的空泡肌病。患者之兄既往有类似肌无力病史,肌肉组织活检未见明显特异性改变,也反映出该病较少累及股四头肌的特点。需注意与以下疾病相鉴别:(1)包涵体肌炎:常见于老年人,偶见家族性发病。临床特点为缓慢发病的下肢近端和上肢远端肌无力,以及吞咽困难,主要累及股四头肌和远端指屈肌,骨骼肌组织活检可见典型空泡肌纤维和炎性细胞浸润,超微结构观察可见管丝状包涵体,可资与遗传性包涵体肌病相鉴别。该例患者为青年男性,病变累及双下肢远端,以胫骨前肌受累为主,病情缓慢进展,且有阳性家族史,肌肉组织活检未见炎性细胞浸润,故不支持包涵体肌炎的诊断。(2)Miyoshi远端型肌营养不良症(MM):亦称青年早发性远端肌病II。该病呈常染色体隐性遗传,致病基因为定位于2p12-14的dysferlin基因,其发病年龄为10-38岁,临床表现为双下肢屈肌对称性肌无力和肌萎缩,尤以腓肠肌受累最为突出,难以足尖站立,亦可出现双上肢和手无力,逐渐累及四肢、面肌和咽部肌等,血清肌酸激酶显著升高,肌肉组织活检显示肌纤维Dysferlin蛋白缺乏,无镶边空泡和包涵体,可与遗传性包涵体肌病相鉴别。该例患者以双侧胫骨前肌受累为主,血清肌酸激酶轻度升高,肌肉组织活检可见明显镶边空泡,故不支持Miyoshi远端型肌营养不良症的诊断。(3)Laing型远端肌病:亦称为青年早发性远端肌病III。呈常染色体显性遗传,系定位于14q11的肌球蛋白重链7(MyH7)基因突变所致,通常于青少年期发病,病情进展缓慢,以胫骨前肌受累显著,可出现严重的足下垂,亦可累及近端肌群,甚至有时可见震颤、周围神经病和心肌炎,血清肌酸激酶正常或轻度升高,肌肉组织活检无典型的镶边空泡和包涵体,可与遗传性包涵体肌病相鉴别。本文病例发病年龄较晚,临床未见震颤、周围神经病和心肌炎等,双侧股四头肌无明显受累,肌肉组织活检可见明显的镶边空泡,基因检测GNE基因突变,故不支持Laing型远端肌病的诊断。 讨论 遗传性包涵体肌病性GNE肌病系GNE基因突变引起的以股四头肌不受累为特点的远端肌病,呈常染色体隐性遗传,亦称为伴镶边空泡的远端肌病、股四头肌不受累的空泡肌病或遗传性包涵体肌病2型(hIBM2)。GNE基因突变使一些重要的糖蛋白或糖脂无法唾液酸化,可能是主要的致病机制,迄今已发现超过150种GNE基因突变亚型。 1981年,日本学者Nonaka等首次报告伴镶边空泡的远端肌病;1984年,Argov和Yarom在中东犹太人种中报告股四头肌不受累的空泡肌病;1995年,Askanas和Engel对类似骨骼肌疾病进行总结,统一命名为“遗传性包涵体肌病”;1996年,Mitrani⁃Rosenbaum等采用全基因连锁分析(genomewide linkage analyses)将呈常染色隐性遗传的遗传性包涵体肌病的致病基因定位于9p1-q1; 进入21世纪,Eisenberg等和Kayashima等确定遗传性包涵体肌病2型和Nonaka肌病的致病基因为GNE基因。 至此,遗传性包涵体肌病2型的致病基因被确定,但是其不同历史名称仍沿用至今,给临床和科研造成不便。直至2014年,经Huizing等建议方将其统一命名为GNE肌病。 GNE肌病在世界范围的患病率约为1/100万。通常于青年发病,早发性和晚发性病例均少见,典型临床症状以缓慢进展的下肢远端肌无力和肌萎缩为主,疾病早期表现为双侧足背屈无力,进行性加重,逐渐累及前臂屈肌、腰带肌、肩带肌甚至中轴肌群,不累及眼外肌、喉部肌、呼吸肌和心肌,高级皮质功能、脑神经、感觉系统和共济运动正常,随着疾病进展,严重累及肩带肌,但三角肌、肱二头肌和肱三头肌受累相对较轻;后期出现下肢远端和近端肌无力,而股四头肌肌力保留完好,下肢近端肌和上肢肌肉缓慢进展,保证患者可依靠髋部结构而长期保持独立行走能力。大部分患者股四头肌肌力相对保留数十年,而少数(5%)患者有不同程度的早期股四头肌无力。目前,股四头肌肌力保留的病理生理学机制尚不十分清楚,有学者认为同一基因突变在同一种组织中的不同部位表现出明显的异质性,可能与体内存在某种补救机制相关,部分肌群中存在与GNE蛋白功能相同的酶,故选择性不累及某些肌群。 辅助检查方面,GNE肌病患者血清肌酸激酶轻至中度升高。肌电图以肌源性损害为主,伴神经源性损害,可能是由于在维持神经肌肉接头(NMJ)稳定性中起重要作用的神经细胞黏附因子(NCAM)低唾液酸化所致。双下肢MRI的典型表现为,早期胫骨前肌、股二头肌和大腿内收肌联合受累,股四头肌相对保留或仅轻度受累,至晚期股四头肌仅股外侧肌不受累。研究证实,双下肢MRI显示同时累及半膜肌、半腱肌和胫骨前肌者,更倾向于GNE肌病的诊断。 骨骼肌组织活检典型表现为HE染色和MGT染色在空泡边缘可见嗜碱性颗粒,而无炎性细胞浸润。这些镶边空泡酸性磷酸酶活性增高,溶酶体标志蛋白呈阳性反应,电子显微镜下可见多层体,提示空泡实质为自噬空泡,这在正常肌纤维中几乎检测不到。电子显微镜观察可见肌纤维核内细丝状包涵体。值得注意的是,镶边空泡并非GNE肌病所特有,亦可见于多种肌肉病,如结蛋白病、其他类型的遗传性包涵体肌病、进行性肌营养不良(PMD)和多种代谢性肌肉病,应注意鉴别诊断。 目前,GNE肌病尚无有效的治疗方法,仅能通过缓解主要临床症状以改善生活质量,包括康复训练、心理支持、并发症的预防与治疗等。外源性补充唾液酸或其中间产物,以及基因治疗是否可以治疗GNE肌病,尚待进一步探讨。 中国现代神经疾病杂志2019年9月第19卷第9期 作者:吴昊 石志鸿 纪勇 ( 天津市环湖医院神经内科) 韩彤( 天津市环湖医院神经影像科)阎晓玲( 天津市环湖医院病理科) 中国现代神经疾病杂志2019年9月第19卷第9期 作者:吴昊 石志鸿 纪勇 ( 天津市环湖医院神经内科) 韩彤( 天津市环湖医院神经影像科)阎晓玲( 天津市环湖医院病理科) 《神外世界》视频号重磅上线 返回搜狐,查看更多 |

【本文地址】