| 一些生物信息学常用的分析法的介绍 | 您所在的位置:网站首页 › 生物学分析软件有哪些名称 › 一些生物信息学常用的分析法的介绍 |
一些生物信息学常用的分析法的介绍
|
我个人认为生物信息学是生命科学和计算机科学还有统计学所构成的一门交叉学科。私以为目前网络上的文献有些晦涩难懂。为了更好地帮助新手入门,现将目前网络上的各种文献资料总结为本文,供各位同行参阅。
目前生物信息学常用的分析法有如下几种: 基因差异表达的显著性分析(又称差异表达分析) DEG功能富集分析(Go分析和Kegg分析)加权基因共表达网络分析(Weighted Gene Co-expression Network Analysis )我们逐次讲起,首先来讲讲基因差异表达的显著性分析。这种分析法又被简称为差异表达分析,通常指的是一个基因在两个条件中表达水平的检测值,再排除实验检测等因素外,达到的一定差异。这个差异既具有统计学意义同时也具有生物学意义。 对于差异表达分析来说常用的算法有算法有三大类: 倍数分析-计算每一个基因在某种两情况下的比值若大于某一定值则为差异表达基因统计模型-t检验、方差分析等方法,计算置信度选取一定P值以下的作为差异表达基因机器学习-贝叶斯,SVM,随机森林等算法倍数分析法由于过于简单粗暴,本文不进行集中介绍。基本上,统计课上都会介绍如何使用T检验来从统计学意义上比较两个样本间的差异,然后在样本量较大的时候考虑使用方差分析。不过这样做的前提是样本来自服从正态分布的群体。这种思想在生物信息学中依然可用。 单总体检验的T值为
其中 在做完差异表达分析之后,你得到了好多P值很小的基因。 下面你可以做功能富集分析来让你的结果更可信。 那么什么是功能富集?功能富集我个人认为就是分类,而分类的标准就是按照基因的功能的不同。为了解决这种分类问题,科学家们联合起来开发了很多数据库。 比较有名的是The Gene Ontology Consortium和Kyoto Encyclopedia of Genes and Genomes,前者通常用于描述基因间的层级关系,而后者大多数人把它当做一个基因通路的数据库,其实不然KEGG是一个整合了基因组、化学和系统功能信息的综合数据库。功能富集分析的算法很多,工具也很多。比如说DAVID,GESA,GoMiner等其中最常用也是最权威的工具便是DAVID。 最后我们来讲讲最近很火的WGCNA加权共表达网络分析。WGCNA适合用于非常复杂的数据,推荐5组以上比如说: 不同器官、组织类型的发育调控;统一组织不同时期的发育调控;非生物胁迫不同时间点的应答;病原物侵染后不同时间的应答;从方法上来讲,WGCNA分为表达量聚类分析和表型关联两部分,主要包括基因之间相关系数计算、基因模块的确定、共表达网络、模块与性状关联四个步骤。 首先计算任意两个基因之间的相关系数。为了衡量两个基因是否具有相似表达模式,一般需要设置阈值来筛选,高于阈值的则认为是相似的。但是这样如果将阈值设为0.8,那么很难说明0.8和0.79两个是有显著差别的。因此,WGCNA分析时采用相关系数加权值,即对基因相关系数取N次幂,使得网络中的基因之间的连接服从无尺度网络分布,这种算法更具生物学意义。第二步通过基因之间的相关系数构建分层聚类树,聚类树的不同分支代表不同的基因模块,不同颜色代表不同的模块。基于基因的加权相关系数,将基因按照表达模式进行分类,将模式相似的基因归为一个模块。这样就可以将几万个基因通过基因表达模式被分成了几十个模块,是一个提取归纳信息的过程。得到模块之后可以进行模块功能富集,模块与形状间的相关性的分析等等。
|
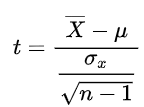
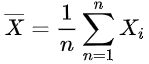 为样本平均数,
为样本平均数,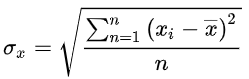
【本文地址】
| 今日新闻 |
| 推荐新闻 |
| 专题文章 |