| 零基础入门转录组分析 | 您所在的位置:网站首页 › 测序的reads是什么意思 › 零基础入门转录组分析 |
零基础入门转录组分析
|
零基础入门转录组分析——第四章(序列比对)
目录
零基础入门转录组分析——第四章(序列比对)1. 之前章节结果的查看1. 构建参考基因组索引2. 序列比对3. 压缩和排序XXX.sam文件4. 构建bam文件的索引(可选)5. 进阶
(我这里使用的虚拟机是vmwarewokstation,版本16.0.0, linux系统是ubantu64位,版本20.04.3)
上一章我们做了质控分析,并对数据进行了质量过滤,这一章序列比对将会用到质量过滤后的数据(02_clean_data)以及第二章准备的参考基因组文件。
本实验选用的是模式生物——C57BL/6J小鼠。
实验分组:药物处理组,对照组,每组6只鼠。
软件:hisat2, samtools
1. 之前章节结果的查看
在前两章中,分别做了原始数据的准备和质控以及质量过滤。 首先来看一下现有的文件和之前的输出结果如下图所示:(质控的结果我给删了,有点占地方)
有了clean_data和准备好的参考基因组,我们接下来第一步就是构建参考基因组索引,要用到的软件是hisat2,如果没有安装过这个软件的同学可以参考软件的安装。 (1)先将01_ref文件夹中的参考基因组压缩包(XXX.fa.gz)和注释文件压缩包(XXX.gtf.gz)解压 gunzip Mus_musculus.GRCm39.dna.primary_assembly.fa.gz 解压参考基因组 gunzip Mus_musculus.GRCm39.108.gtf.gz 解压参考基因组注释文件 解压后的结果如下图所示:(xxx.fa文件就是我们要用到参考基因组) 先通过pwd指令,查看当前文件夹的路径: hisat2-build指令不难,hisat2-build后跟的是参考基因组的路径,再后面是输出结果的路径,我这里是输出到当前文件夹下,命名为genome…,再后面跟的是hisat2-build结果输出报告(hisat2-build.log 文件) 构建参考基因组索引这步会很慢,需要耐心等待 结果如下图所示,多了一些白色的genome…的文件,这些就是构建好的参考基因组索引文件(注意备份) 构建好索引文件后,接下来就要将过滤后的原始数据比对到参考基因组上,首先我们先了解一下常用的参数: -new-summary 这会让hisat2软件比对后输出的报告会更加好看-p 是线程数,我的虚拟机是内存8G,分配了4个处理器,256G硬盘空间,线程数跟处理器的数量有关,这个线程数设的越大,运行速度会越快(根据你的虚拟机状况选用合适的线程数)。-x 这里输入构建好的参考基因组索引的路径-1 输入过滤后的原始数据1的路径-2 输入过滤后的原始数据2的路径-S 输出的XXX.sam文件-rna-strandness 这个参数是链特异性文库需要的,就是说你要问测序公司你的原始数据是普通文库数据还是链特异性文库。如果是普通文库,这里就不加-rna-strandness这个参数(hisat2软件会比对两次,正向一次,反向互补的链再比对一次);如果是链特异性文库,就要加上-rna-strandness参数RF 参数代表的是双端测序,如果是单端测序,这里就是R首先在桌面路径下通过mkdir 03_hisat2_result创建一个03_hisat2_result文件夹(用来存放比对结果) 接下来看代码(我这里用的全是绝对路径,绝对路径好处就是你在任何路径下运行代码都没问题,但是缺点就是特别长,会看花眼) hisat2 --new-summary -p 2 -x /home/daizhuer/Desktop/01_ref/genome -1 /home/daizhuer/Desktop/02_clean_data/A2_1_val_1.fq.gz -2 /home/daizhuer/Desktop/02_clean_data/A2_2_val_2.fq.gz -S /home/daizhuer/Desktop/03_hisat2_result/A2.sam --rna-strandness RF 1>/home/daizhuer/Desktop/03_hisat2_result/A2.log 2>&1 注意:hisat2比对生成的XXX.sam文件会非常大,大概20G左右,所以一定要留出足够的空间 运行结果如下,我们可以看到生成了两个文件,一个是20G的A2.sam文件,另一个是A2.log文件,这里存放比对结果的分析报告。 通过cat A2.log指令可以查看比对结果分析报告,如下图所示: 根据前面的图,我们可以看到sam文件非常大,这么大的数据不利于保存和分析,现需要对其进一步压缩和排序。 这里我们需要用到samtools软件(如果没安装的小伙伴可以通过conda install samtools -y指令安装即可)samtools软件可以轻松对sam文件进行压缩和排序,输出结果为XXX.bam文件,指令如下: sort 参数表示进行排序-o 是输出文件的路径samtools sort -o /home/daizhuer/Desktop/03_hisat2_result/A2.bam /home/daizhuer/Desktop/03_hisat2_result/A2.sam 代码大致意思就是输入03_hisat2_result文件夹中的A2.sam文件,通过samtools软件对其压缩和排序,输出结果到03_hisat2_result文件夹下命名为A2.bam。 结果如下图所示: 注意:XXX.log文件不要删,这可以帮助后期查看比对率 4. 构建bam文件的索引(可选)有了bam文件,接下来可以对bam文件构建索引,构建索引是为了能够在IGV软件中观察比对结果,这步不是必须的,可以根据需求选择性观看。 构建bam文件的索引非常简单,通过samtools index A2.bam指令就能完成。 结果如下: 在这一部分,主要介绍一下如何批量比对 (1)准备样本信息表 切换到02_clean_data文件夹路径下 接下来通过vim sample_ifo指令编辑文件,如下图所示,是刚开的样子 编辑好后摁下Esc,输入:wq保存并退出。 (2)脚本编辑 有了样本信息表之后,我们就可以开始写脚本了,先切回到03_hisat2_result文件夹目录下,输入如下指令: awk '{print "hisat2 --new-summary -p 2 -x /home/daizhuer/Desktop/01_ref/genome -1 /home/daizhuer/Desktop/02_clean_data/"$1" -2 /home/daizhuer/Desktop/02_clean_data/"$2" -S /home/daizhuer/Desktop/03_hisat2_result/"$3".sam --rna-strandness RF 1>/home/daizhuer/Desktop/03_hisat2_result/"$3".log 2>&1"}' /home/daizhuer/Desktop/02_clean_data/sample_ifo > run_hisat2.sh输出为一个名为run_hisat2.sh的脚本文件 代码较长,你们可以粘贴下来慢慢理解,其实和序列比对那里的代码很像,只不过有一些需要用到名称的地方用刚才准备的样本信息表中不同列的名称给代替了。 例如: hisat2 --new-summary -p 2 -x /home/daizhuer/Desktop/01_ref/genome -1 /home/daizhuer/Desktop/02_clean_data/"$1(这个$1就是样本信息表中的第一列,也就是刚才样本信息表中的A2_1_val_1.fq.gz,后面的$2…依次类推) 如果你的样本信息表有多行,那么生成的这个XXX.sh脚本每一行都有这样相似代码,只不过分析的样本名称不同。 脚本生成之后就可以通过bash XXX.sh命令运行了,不过这里一旦运行就要保证有足够的空间,因为一次比对就会生成20g的文件,需要保证足够的空间。 压缩和排序的自动化也类似,用awk指令即可拼接出一个简单脚本。 awk ‘{print “…”}’ 样本信息表 > XXX.sh 注意:如果这里理解的比较困难可以查看第二章质控进阶那部分,那里awk代码较短,更容易理解。 以上就是质控的所有过程,如果有什么需要补充或不懂的地方,大家可以私聊我或者在下方评论。 后续的干货内容会逐渐更新,如果觉得本教程对你有所帮助,多多点赞加关注昂,我们下期再见!!! |
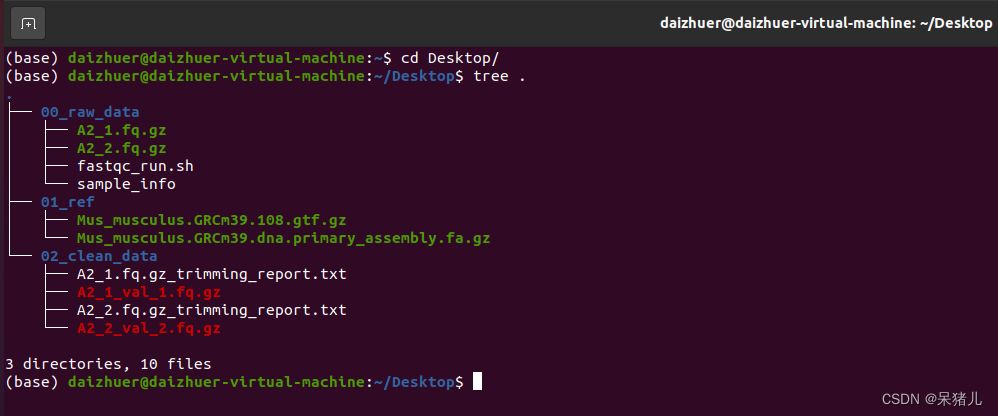 00_raw_data文件夹中是我们的原始数据和样本信息表,但是有了clean_data这些就用不到了。 01_ref文件夹中有参考基因组(XXX.fa.gz)和注释文件(XXX.gtf.gz) 02_clean_data文件夹中有上一章做完质量过滤的数据(又叫做clean data)。
00_raw_data文件夹中是我们的原始数据和样本信息表,但是有了clean_data这些就用不到了。 01_ref文件夹中有参考基因组(XXX.fa.gz)和注释文件(XXX.gtf.gz) 02_clean_data文件夹中有上一章做完质量过滤的数据(又叫做clean data)。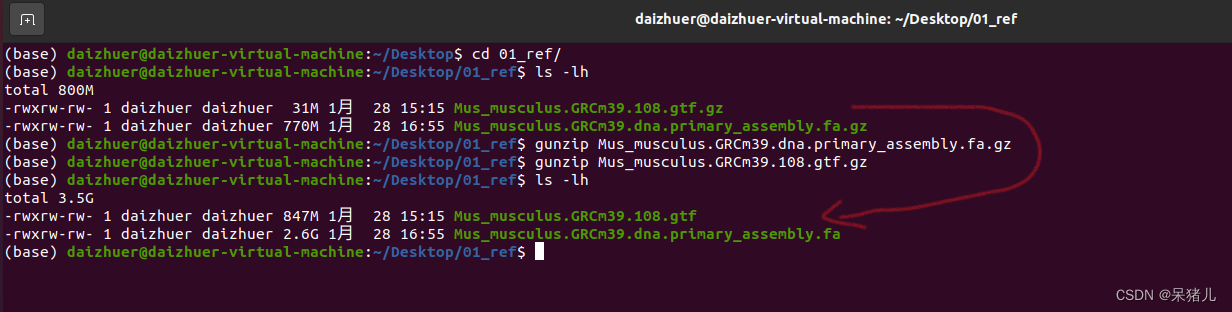 (2)接下来是构建参考基因组索引文件:
(2)接下来是构建参考基因组索引文件: 接下来用hisat2-build 构建参考基因组的索引:(我这里用的是绝对路径) hisat2-build /home/daizhuer/Desktop/01_ref/Mus_musculus.GRCm39.dna.primary_assembly.fa /home/daizhuer/Desktop/01_ref/genome 1>hisat2-build.log 2>&1
接下来用hisat2-build 构建参考基因组的索引:(我这里用的是绝对路径) hisat2-build /home/daizhuer/Desktop/01_ref/Mus_musculus.GRCm39.dna.primary_assembly.fa /home/daizhuer/Desktop/01_ref/genome 1>hisat2-build.log 2>&1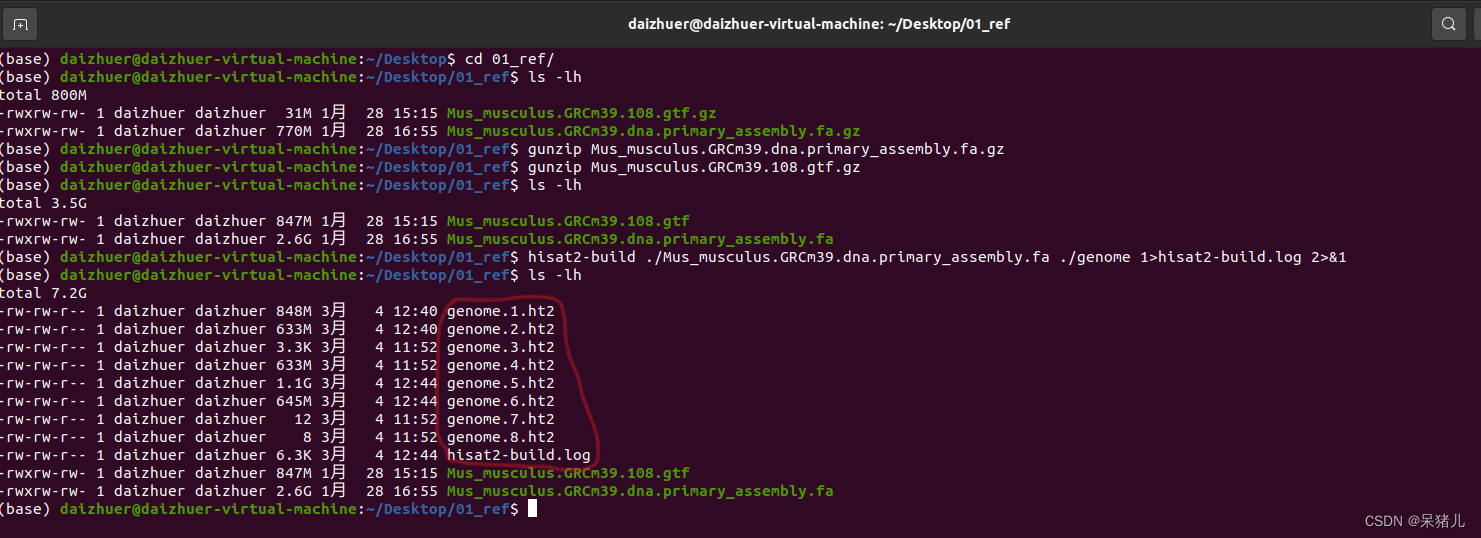
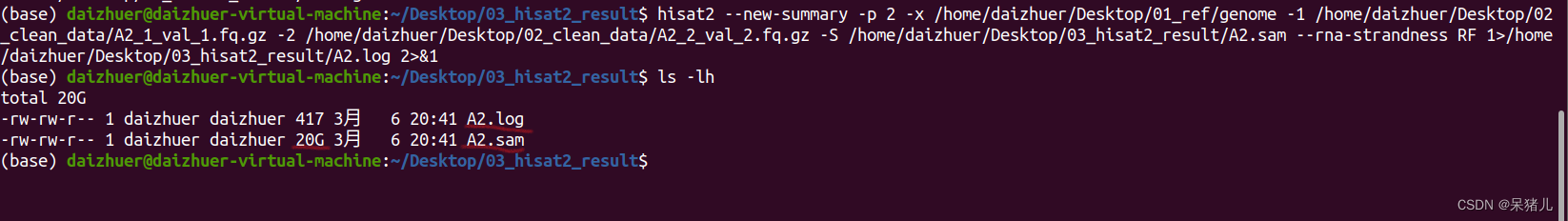
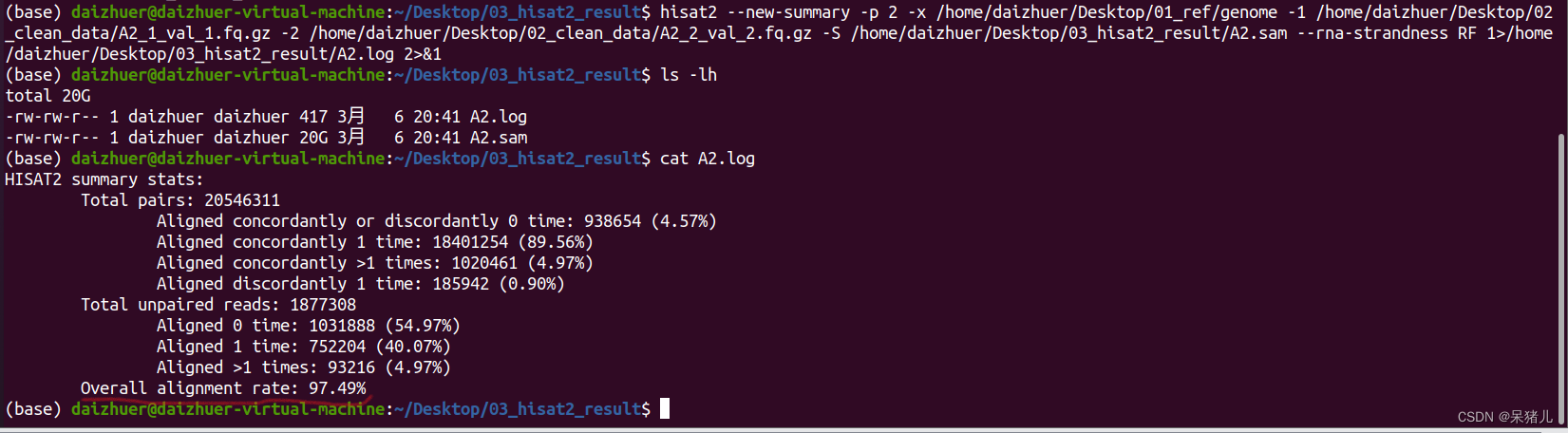 结果显示97.49%的reads都比对到参考基因组了(说明比对结果非常好!)
结果显示97.49%的reads都比对到参考基因组了(说明比对结果非常好!)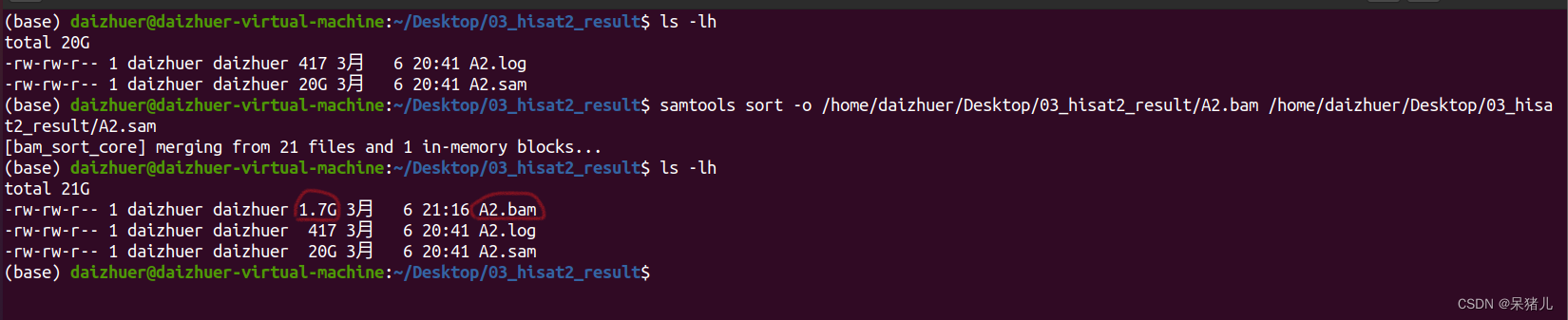 压缩和排序后生成了一个1.7G的A2.bam文件,相比于sam文件,小了10倍,有了bam文件,sam文件就没啥用了,可以删除节省空间(如果电脑或移动硬盘比较大的同学,可以选择两个都备份)。
压缩和排序后生成了一个1.7G的A2.bam文件,相比于sam文件,小了10倍,有了bam文件,sam文件就没啥用了,可以删除节省空间(如果电脑或移动硬盘比较大的同学,可以选择两个都备份)。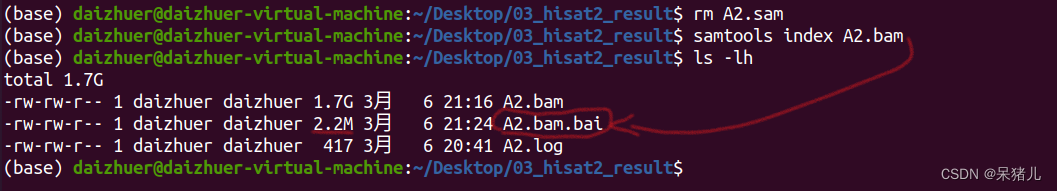 生成了一个2.2Mb的XXX.bai文件,这就是索引文件,通常是在IGV软件中使用,可视化比对结果。
生成了一个2.2Mb的XXX.bai文件,这就是索引文件,通常是在IGV软件中使用,可视化比对结果。 输入指令:ls *fq.gz > sample_ifo,会在当前路径下生成一个sample_ifo文件
输入指令:ls *fq.gz > sample_ifo,会在当前路径下生成一个sample_ifo文件 点一下键盘上的i键进入编辑模式,修改成下图所示的样子:
点一下键盘上的i键进入编辑模式,修改成下图所示的样子: 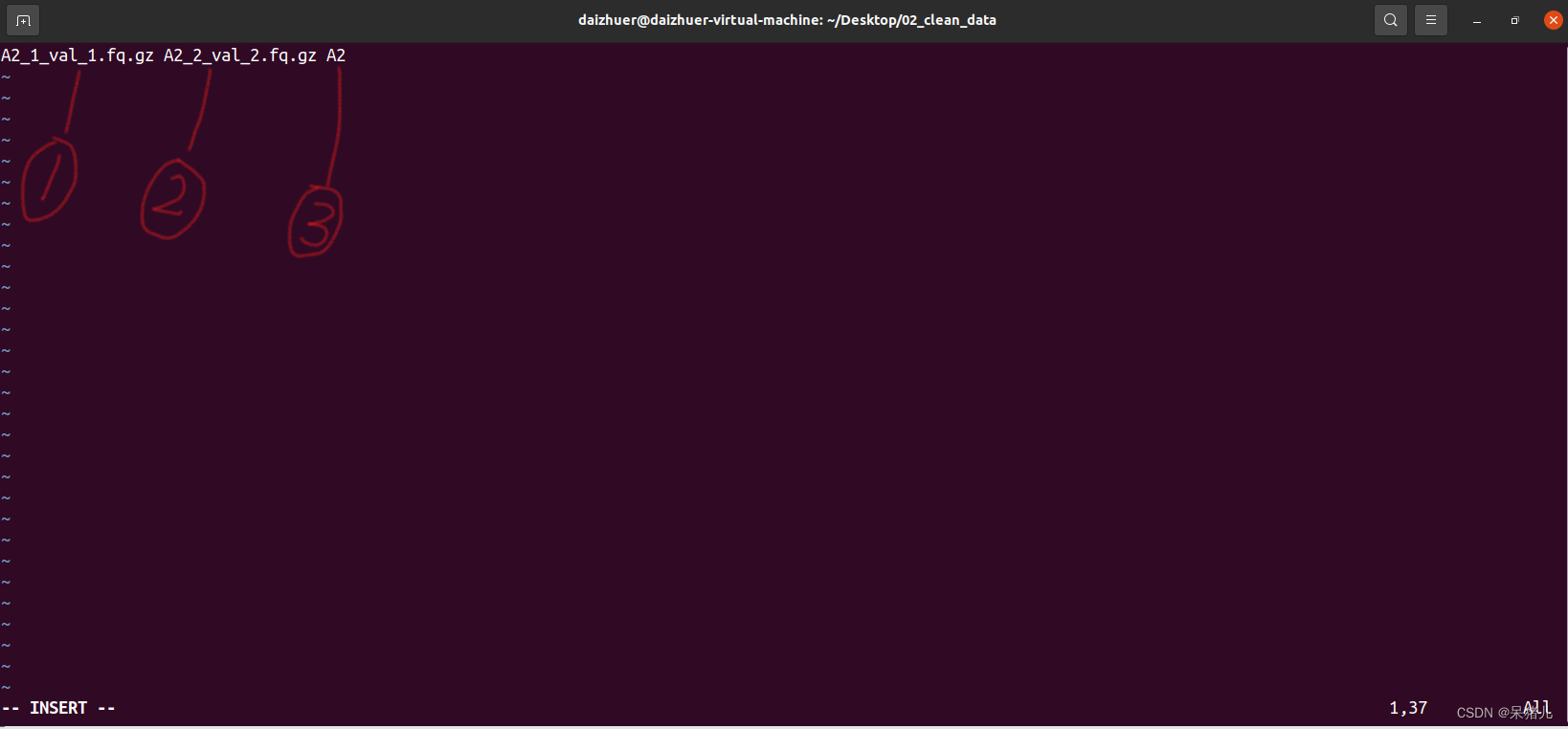 中间以空格相隔,一共三列,前两列就是clean_data的名称,第三列是你想要的样本名(可以随便取,自己记得是啥就行,记住这三列,等会会用到)
中间以空格相隔,一共三列,前两列就是clean_data的名称,第三列是你想要的样本名(可以随便取,自己记得是啥就行,记住这三列,等会会用到)【本文地址】