| scRNA分析 | 您所在的位置:网站首页 › 小提琴放置 › scRNA分析 |
scRNA分析
|
单细胞常见的可视化方式有DimPlot,FeaturePlot ,DotPlot ,VlnPlot 和 DoHeatmap几种 ,Seurat均可以实现,但文献中的图大多会精美很多。比如 惊艳umap图: scRNA复现|所见即所得,和Cell学umap,plot1cell完成惊艳的细胞注释umap图; DimPlot美化 scRNA分析 | 定制 美化FeaturePlot 图,你需要的都在这, DotPlot美化scRNA分析| 和SCI学 定制化聚类点图(Dotplot ),含二行代码出图方式, DoHeatmap 热图:scRNA分析| DoHeatmap 美化,dittoSeq ,scillus 一行代码出图,你PICK谁? 本次介绍Seurat 以及 ggplot2绘制,优化堆叠小提琴图的方法。 一 载入R包,数据 仍然使用之前注释过的sce.anno.RData数据 ,后台回复 anno 即可获取。  二 Seurat 调整,美化 1,基础VlnPlot图 首先计算marker基因,然后使用seurat的DoHeatmap 函数绘制初始热图 当展示少量基因时候,很清晰 。但是更常见的时候需要同时展示各个cluster/celltype的marker gene ,这时候就会看不清晰。  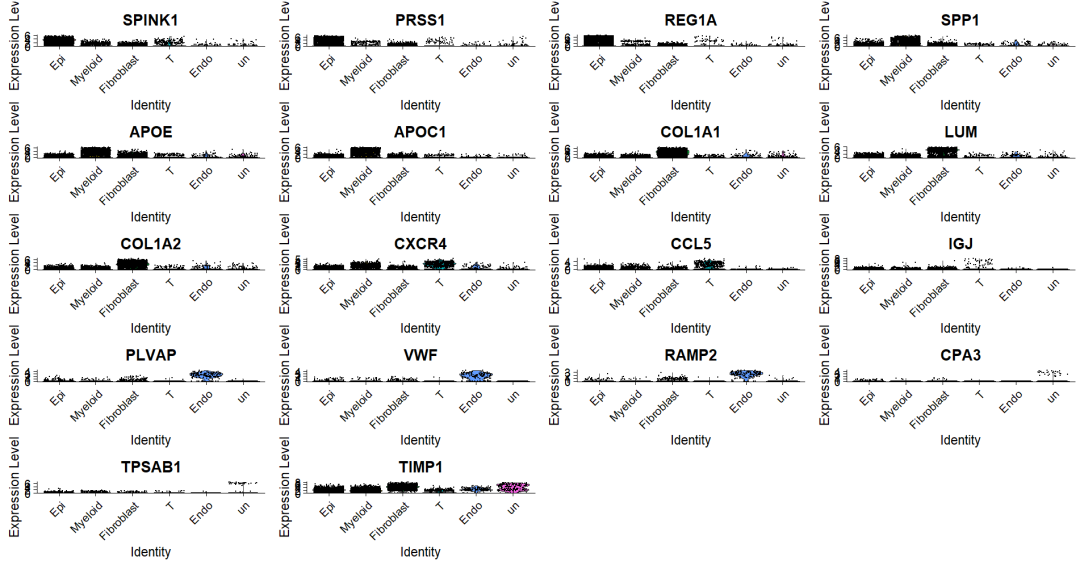 2,Seurat-堆叠VlnPlot图 2,Seurat-堆叠VlnPlot图Seurat的VlnPlot函数中stack 参数可以实现堆叠小提琴图,flip 是否翻转 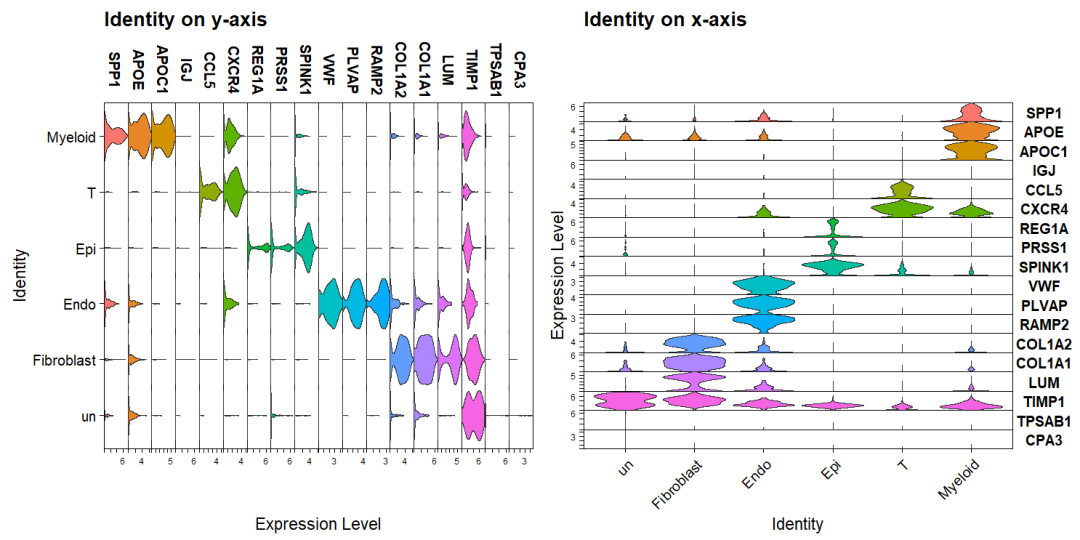 3,Seurat-优化颜色,大小,方向 3,Seurat-优化颜色,大小,方向自定义颜色,是否排序,主题等信息更是和前面的一样,直接添加theme信息即可。 注意如果想要每种cluster/celltype是一种颜色的话使用split.by参数。 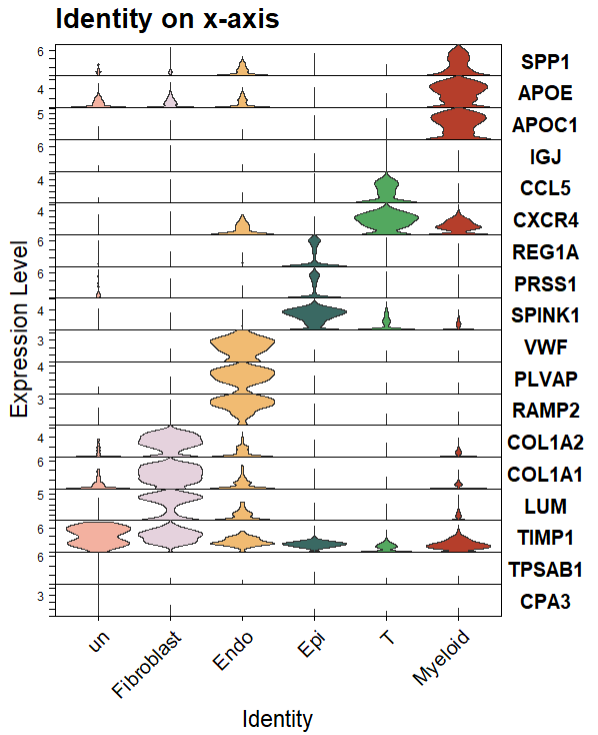 Seurat的堆叠小提琴图其实已经可以了,当然也可以使用ggplot2进行更多的自定义。 三 ggplot2-堆叠小提琴图 1,提取,转化数据首先使用FetchData提取出marker gene的表达量,celltype /seurat_clusters(宽数据),然后转为ggplot2读取的长数据类型 。 此外对照上述的图,可以看到celltype /seurat_clusters一个表达量值,而FetchData得到的是每个cell 的表达量,因此还需要计算每种cluster的基因均值。  2,ggplot2 绘制-核心 2,ggplot2 绘制-核心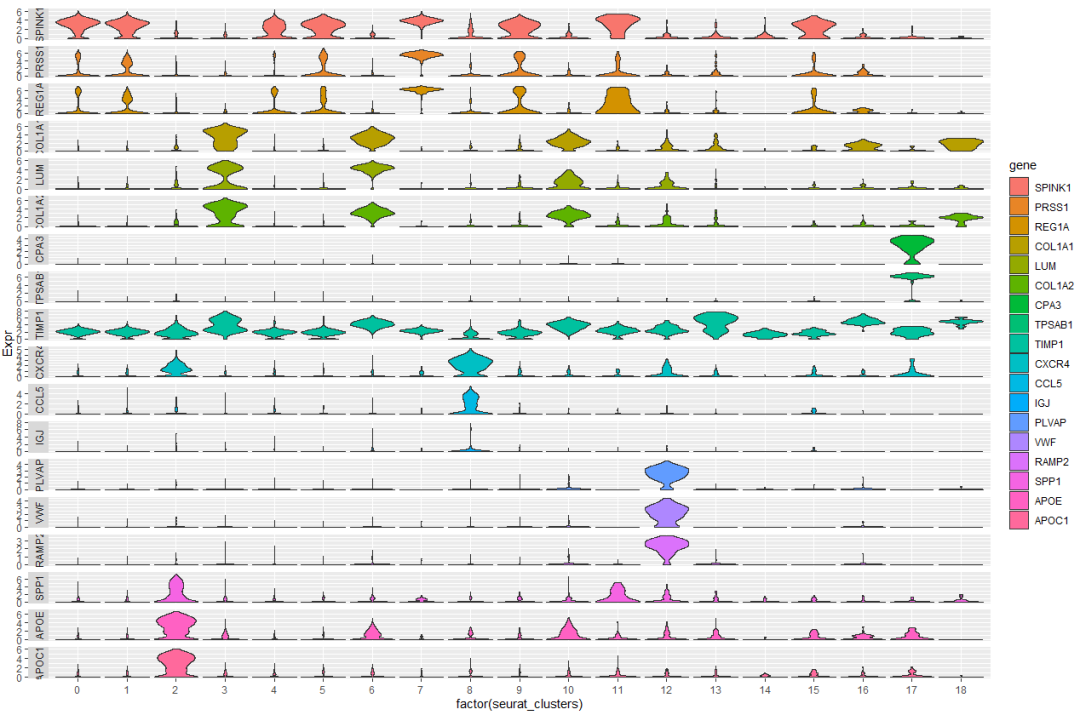 3,ggplot2 绘制-优化 3,ggplot2 绘制-优化上述是ggplot2绘制堆叠小提琴图的核心代码,可以做很多调整 (1)主题(大小,颜色),legend 等 (2)“翻转”(使用aes调整横纵坐标)  (3)添加基因的分组/注释 A:添加分组,注释 假设知道marker gene的通路,也可以添加上(为了美观先隐藏p1中的横坐标基因标签) B:构建注释信息-基因分组信息 这里通路是随便写的,仅为示例,并不是该marker gene 在的通路。 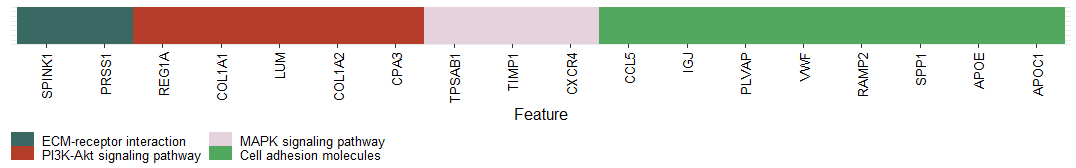 C:拼图收工  参考资料: https://github.com/ycl6/StackedVlnPlot |
【本文地址】