| RNA | 您所在的位置:网站首页 › 如何获取sam文件的内容 › RNA |
RNA
|
对测序得到的reads进行计数,即基因表达的定量过程。根据reads和基因位置的overlap,以此来判断reads到底属于哪一个基因,同时对该reads总数进行计数,生成counts矩阵。 今日内容 1.HTSeq-count对reads进行计数 2.R语言完成counts矩阵的合并1. HTSeq-count对reads进行计数 首先了解HTseq用法,参数说明如下: usage: htseq-count [options] alignment_file gff_file positional arguments: samfilenames Path to the SAM/BAM files containing the mapped reads. If '-' is selected, read from standard input featuresfilename Path to the file containing the features optional arguments: -h, --help show this help message and exit -f {sam,bam}, --format {sam,bam} type of data, either 'sam' or 'bam' (default: sam) 输入文件类型sam/bam;默认为sam文件 -r {pos,name}, --order {pos,name} 'pos' or 'name'. Sorting order of (default: name). Paired-end sequencing data must be sorted either by position or by read name, and the sorting order must be specified. Ignored for single- end data. 输入文件的排序方式,默认按read名排序 --max-reads-in-buffer MAX_BUFFER_SIZE When is paired end sorted by position, allow only so many reads to stay in memory until the mates are found (raising this number will use more memory). Has no effect for single end or paired end sorted by name -s {yes,no,reverse}, --stranded {yes,no,reverse} whether the data is from a strand-specific assay. Specify 'yes', 'no', or 'reverse' (default: yes). 'reverse' means 'yes' with reversed strand interpretation -a MINAQUAL, --minaqual MINAQUAL skip all reads with alignment quality lower than the given minimum value (default 10) 剔除mapping quality值低于阈值的read -t FEATURETYPE, --type FEATURETYPE feature type (3rd column in GFF file) to be used, all features of other type are ignored (default, suitable for Ensembl GTF files: exon)前一期利用SAMtools工具对比对产生的sam文件进行格式转换并排序,得到了按染色体位置进行排序的bam文件${id}.sorted.bam。
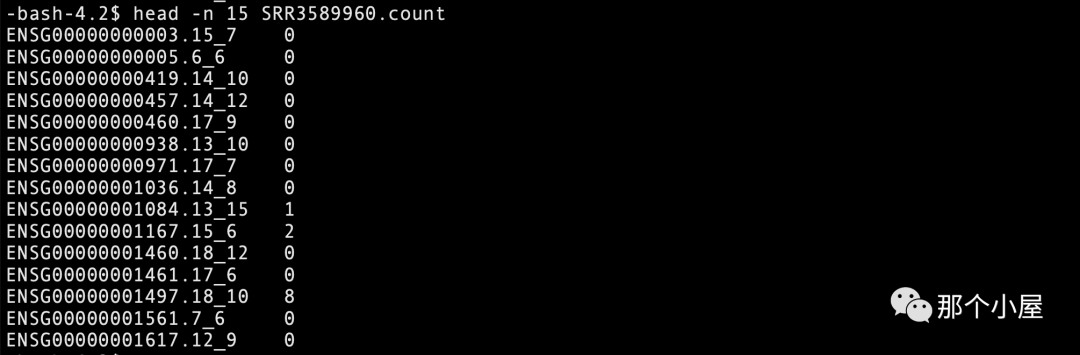
因此输入文件格式-f bam,文件名${id}.sorted.bam ;排序方式-r pos ;文献中提到reads计数时剔除了mapping quality < 30的低质量reads,即-a 30。 输入bam文件路径:./aligned/${id}.sorted.bam 参考基因组gtf索引文件./genome_gtf/gencode.v41lift3_.gtf 生成的count文件存放于./aligned/ for id in `seq 56 62`; do #bsub -J ${id} -n 8 -o %J.${id}.out -e %J.${id}.err -R span[hosts=1] -q normal \ htseq-count -r pos -a 30 \ -f bam ./aligned/${id}.sorted.bam \ ./genome_gtf/gencode.v41lift37.basic.annotation.gtf >./aligned/${id}.count done运行结束将产生.count结尾的文件,head查看一下count文件前15行,了解一下数据结构。结构是一个二维矩阵,第一列为Ensemble ID,小数点后面的数字部分为ID版本信息;第二列即为reads数,可理解为表达量。
至此基于Linux系统的RNA-seq数据上游分析流程基本完成;下游分析主要是可视化过程,依赖R语言来实现~ 2.基于Rstudio的下游分析——counts矩阵的准备 回观count文件的数据结构,你会发现我们面临一个问题:第一列ENS编号并不是我们所悉的基因名称(gene_id)。因此在下游分析前还需完成Ensemble ID版本号的去除及向Gene_ID的转换,如果是含有多个样本的count矩阵,还需将多个count矩阵合并为一个多维矩阵。以上流程可通过Rstudio来完成,使用的R包包括stringr(str_split )、biomaRt(getBM)、merge函数。 2.1. 软件准备 R包使用逻辑:安装——R包调用——函数调用;对于没有安装的R包可通过'install.package("package name")'命令进行下载安装。但使用以上命令安装biomaRt及其依赖包AnnotationDbi时将报错,无法获取安装包信息,可使用BioMannager进行安装,亲测有效 install.package('stringr‘) BiocManager::install("“biomaRt”") BiocManager::install("AnnotationDbi")#软件包下载 library(biomaRt)#R包调用 library(stringr)2.2.多个count矩阵的合并(merge) 由于这个系列所用测序数据包含7个测序数据(SRR56~62),在reads计数时会各自生成一个count矩阵,下游分析时一个一个处理count文件比较麻烦,需将七个count文件进行合并;由于reads计数时使用的是同一个基因组索引文件,因此生成的count文件Ensemble ID必定是一致的,这就方便我们以第一列ID为交集索引实现文件合并。使用的函数为基础函数merge。
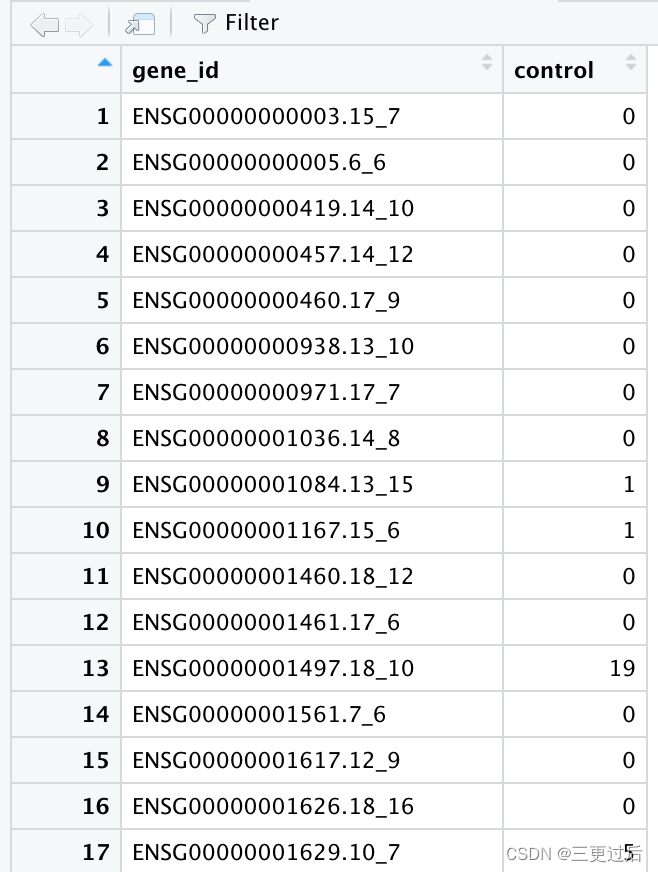
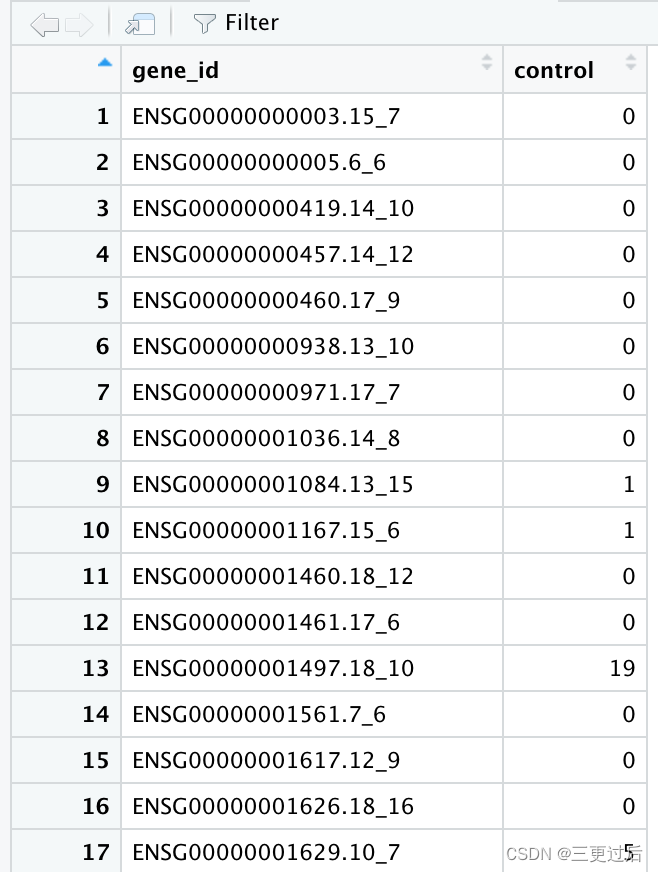
上图为SRR3589960~61 count文件部分行名展示 注⚠️:为节省时间,最初只选用了SRR3589960~SRR3589962三个mRNA测序数据进行测试,因此之后的流程只选择这3个测序数据对应的count矩阵进行演示; 首先通过help('merge')查看函数基本参数用法 #用法Usage: merge(x, y, by = intersect(names(x), names(y)), by.x = by, by.y = by, all = FALSE, all.x = all, all.y = all, sort = TRUE, suffixes = c(".x",".y"), no.dups = TRUE, incomparables = NULL, ...) #x,文件1;y,文件2;by参数对于文件1与文件2的交集列(行)名 #all,如果为 TRUE,x中的每一行在y中没有匹配的行将被添加到输出结果中且这些行通常用NA填充;默认为 FALSE #根据count数据结构,确定使用参数有x,y,by,公共列表头名称gene_id;合并生成名为all_count的文件 all_count |

【本文地址】