| 实用指南丨GSEA详细使用指南与避坑要点 | 您所在的位置:网站首页 › 基因富集分析图横坐标 › 实用指南丨GSEA详细使用指南与避坑要点 |
实用指南丨GSEA详细使用指南与避坑要点
|
现在科研常常会使用到二代高通量测序(NGS),它能获得经过不同处理后实验组和对照组的差异基因,而为了进一步分析差异基因的意义或缩小差异基因的筛选范围,我们经常会根据不同数据库的信息进行富集分析。而富集分析是将基因根据一些先验的知识(即常见的注释信息)进行分类的过程,常见的富集分析有GO/KEGG的超几何富集分析,还有另一种基因富集分析GSEA(gene-set enrichment analysis)。那么,您知道它们的区别在哪里吗? 1、GSEA简介 常规的GO(Gene Ontology)和pathway(KEGG)分析,属于超几何富集算法,使用的基因数据源是我们根据实验组vs对照组所获得的差异基因,其中差异基因则需要根据设置的阈值进行判断,比如p值、FDR值、Fold Change等,因此这就会涉及到人为的阈值选择,具有一定的主观性,毕竟没有一个公认的、固定的阈值标准告诉我们,这样的基因就是差异的基因,所以这种富集结果可能有一定的局限性。 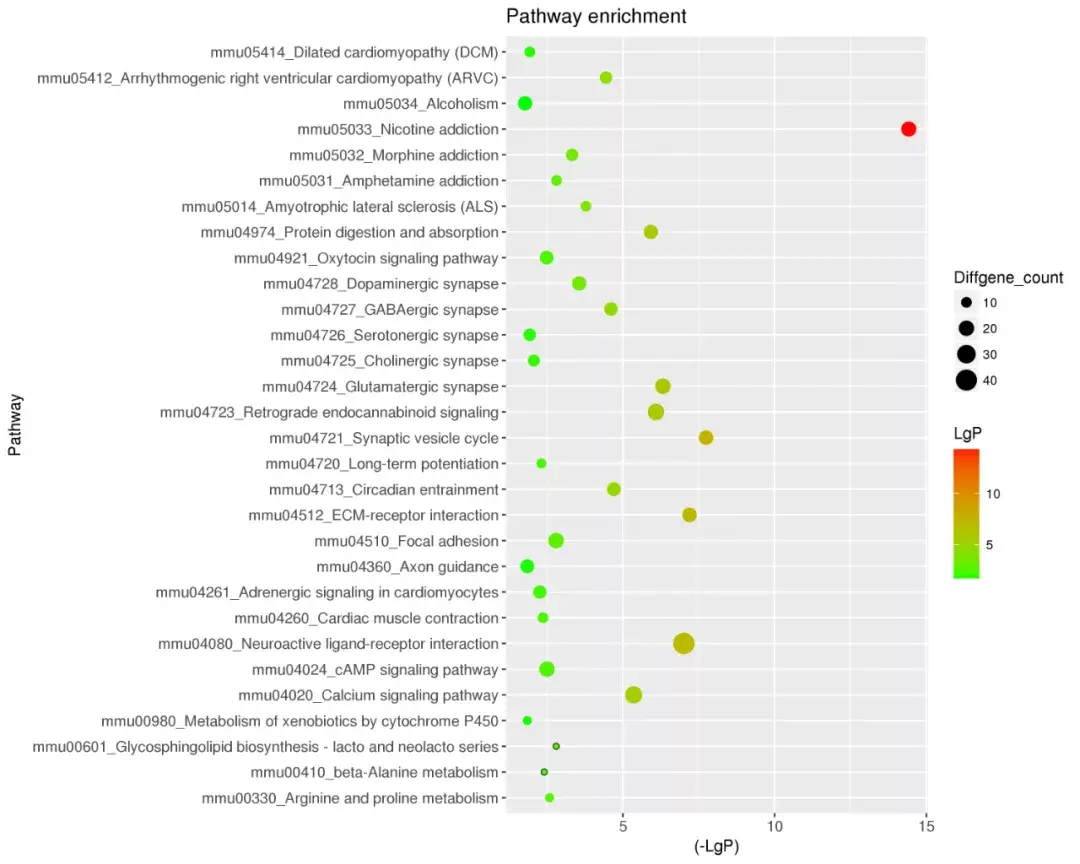 KEGG富集结果之气泡图 基因集富集分析GSEA采用的算法则不同,使用的基因数据源是我们实验组和对照组检测到的所有基因(无论是否被人为阈值判断为差异基因),将所有基因与预定义的GSEA基因集(类似pathway、GO这样的基因与功能对应关系)进行比较、富集,从而判断基因对表型(功能)的贡献。因此,从基因集的富集角度出发,GSEA不局限于是否为差异基因,理论上更容易发现一些对生物通路/功能有细微变化(基因倍数变化小)的影响。  GSEA富集结果 总的来说,二种富集分析都是可以使用的方法,而GSEA涵盖的变化基因更多,哪怕有些基因在我们看来不是”差异基因”,因此,GSEA相对来说要更为全面一些。如果不满足于常规富集的研究者,不妨试试GSEA的富集分析。 那么,今天我们就一起来使用GSEA本地软件,学习GSEA的准备数据、软件使用以及结果解读等过程,帮你快速学会如何进行GSEA分析。 2、GSEA数据准备 首先,要进行GSEA富集分析,我们需要准备几部分的数据:1)预定义的基因集;2)所有样本的所有基因表达矩阵;3)样品的分组信息。接下来,我们将依次讲解各部分的数据内容和要求。 01 预定义的基因集-Gene sets database GSEA基因集来源GSEA基因集就像KEGG中某条pathway中对应哪些基因一样,GSEA基因集也是对基因的注释。每个基因集,一般是我们从其它数据库或权威文献总结获得,比如Pathway注释信息,GO注释信息。在GESA官网(http://www.gsea-msigdb.org/)中,我们可以下载官方提供给我们的基因集,但官方只提供了有限的一些基因集,只有人类的9大类基因集,包括了Pathway、GO、Reactome等,如果我们要做其它物种,如大鼠、小鼠、拟南芥等,就需要自己根据其它物种的已有注释信息,构建可运行的GSEA基因集格式。 PS:如果有必要,研究者完全可以自行根据文献或者其它数据来源,构建一个或多个全新的注释条目,同样可以使用GSEA的方法运行。 GSEA基因集下载现在官方下载的基因集为txt格式(制表符分隔),需要手动更改后缀名为gmt,否则后续加载到软件时会报错。不过官方的人类基因集gmt都可以在本地化软件中在线选取,所以如果做人的富集分析,也无须提前下载。GSEA基因集内容与格式基因集gmt格式为文本数据,数据分隔方式为制表符分隔,可使用Excel打开,打开后我们可以看到数据格式如下,文件没有列头信息:第一列为基因集的名称ID,可看到我们选取的是KEGG的pathway信息用于做富集分析;第二列为对基因集的描述信息,可有可无,但不可删除该列,当无任何描述时,可随意填写文本占位,如全部填写“na”,如果随意删除第二列描述信息,则会导致软件读取后续的注释基因为描述信息,最终每个基因集都会少一个基因,即某基因集有60个基因注释,删除第二列后,经过GSEA软件读取,就只能找到该基因集59个基因注释;第三列以及后续列均为该基因集所包含的具体基因,注意基因的形式,有ENTRZID的数字形式和SYMBOL形式(注意区分大小写),这要求后续输入的表达矩阵的基因形式要匹配,如果基因形式无法对应,则软件无法将输入基因与基因集中基因进行匹配,也就无法注释,最终分析结果会出错。  GSEA gmt基因集格式内容 GSEA基因集下载现在官方下载的基因集为txt格式(制表符分隔),需要手动更改后缀名为gmt,否则后续加载到软件时会报错。不过官方的人类基因集gmt都可以在本地化软件中在线选取,所以如果做人的富集分析,也无须提前下载。 02 全样本的表达矩阵-Expression dataset 常规的二代测序,在测序数据比对基因组之后,都会产生所有样本的所有基因的表达矩阵,通常会产生原始counts表达值,以及某种标准化的表达值(如FPKM、TPM等等),我们也可以在差异结果中找到未经筛选的、包含所有基因的分析结果,选择合适的列数据,用于构建表达矩阵,格式要求行为基因,列为样本,最后导出为制表符分隔的txt即可。 注意1:表达值的选取建议使用分析结果中每个基因的标准化值,因为原始counts不能很好体现组别变化趋势; 注意2:基因名称的形式选择,要与我们的gmt基因集中一致,否则会出错; 注意3:所有基因的表达矩阵需要去除没有表达的基因行,即表达值全为0的;(或根据自己的要求筛选) 注意4:样本名称的顺序,后续会设置分组信息,建议顺序按照组别进行放置;  所有基因的分析结果 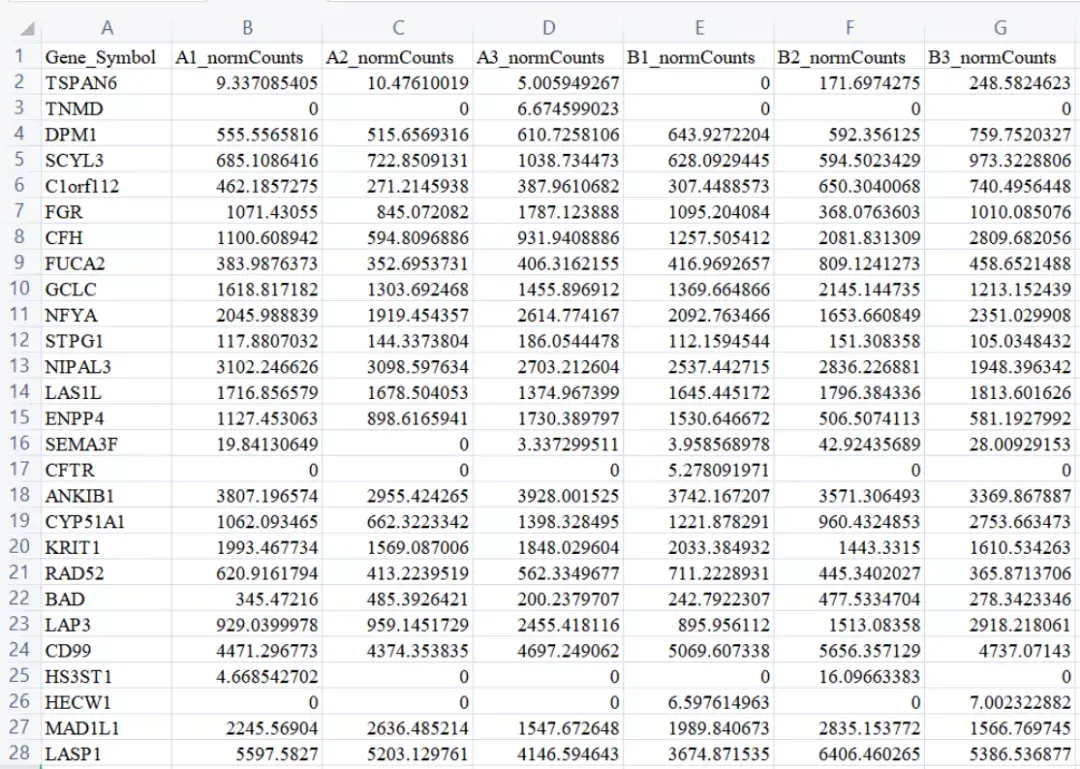 合适的基因表达矩阵 03 分组信息-Phenotype labels 分组信息可在官网下载模板进行修改,也可以在Excel中根据GSEA的要求进行填写,填写完成后,再导出成制表符分隔的txt文件,最后将后缀名改为cls即可。 分组信息要求如下:  第一行:样本总量,分组/类型的数量,以及固定格式”1”;第二行:固定格式”#”,分组的名称1,分组名称2;(分组名称可与样本名称不同,不影响数据读取)第三行:每个样本具体的分组类型,填写的数量一定要与准备的表达矩阵的样本数量对应,且分组顺序和表达矩阵中样本顺序也要对应,因为GSEA软件会根据该分组信息去表达矩阵中提取对应列的表达值进行后续分析,所以一旦写错,富集结果就是错误的。 04 小结 总体来说,GSEA的软件会根据我们提供的分组信息,去表达矩阵中提取与分组相关的表达数据,然后进行组间分析,随后继续进行富集分析时,会将表达矩阵中的基因名称与预设基因集的基因名称进行匹配,最后产生富集结果。 3、GSEA运行 GSEA本地化软件在官网(http://www.gsea-msigdb.org/gsea/downloads.jsp)可以轻松找到下载地址,根据自己的系统选择下载安装,同时软件的使用需要java环境,别忘记安装哦~GSEA的运行,可分为准备数据的加载、运行参数的设置、产出结果三个部分。 01 数据加载-Load data 软件打开后,不能直接RunGSEA,而是需要先Load data,需要先将我们刚才准备的3个文件进行加载(若使用官方的人类预设基因集,则只需要加载其它文件即可),加载正确后有提示,且Object cache会正确显示相关信息。 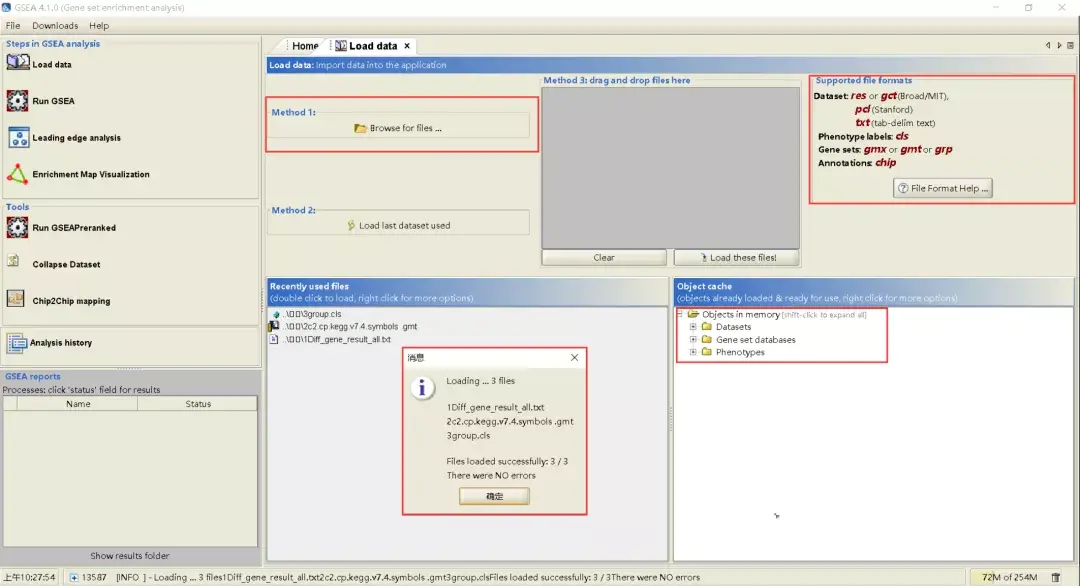 02 数据与参数选择 在Load data后,我们可以点击RunGSEA进入到运行参数设置界面。GSEA包含3部分参数设置,Required fields、Basic fields和Advanced fields,其中Required fields为必须手动填写参数,其余为可选修改项。  Required fields 1)Expressiondataset:选择我们加载的表达矩阵,如果有多表达矩阵时,注意区分清楚;2)Gene stes database:点开下拉菜单后,选择Gene matrix(local gmx/gmt),选择我们刚才加载的基因集(也可以选择从官网website下载数据,即Gene matrix(from website));3)Number ofpermutations:随机置换次数,越大越好,官方通常采用默认设置1000;4)Phenotype labels:选择我们加载的分组,一般选择Exp vs Con;(也可在线建立分组信息);5)Collapse/Remapto gene symbols:非芯片数据则选择“No_Collapse”,表示使用表达矩阵的原始基因ID进行映射到预设基因集(此为常用设置),如果是芯片,则根据需求选择“Collapse”或“ Remap_Only”;6)Permutation type:评估富集得分Enrichment Score的统计显著性时候,执行的排列类型。官方建议每组样本数目大于7个时,建议选择phenotype,否则选择gene sets;7)Chip platform:和上面Collapse/Remap to gene symbols参数对应,选择对应的芯片注释信息(No_Collapse无需选择); Basic fields 1)Analysis name:项目名,可根据需求进行命名;2)Max size:从预设基因集中筛选掉不在表达矩阵中的基因后,预设基因集所包含的基因数量大于此值时,基因集将被排除在分析之外;3)Min size:从预设基因集中筛选掉不在表达矩阵中的基因后,预设基因集所包含的基因数量小于此值时,基因集将被排除在分析之外;4)Save results inthis folder:结果保存路径; #例如预设的基因集有1000个注释基因,去除掉不在表达矩阵中的基因后,还剩下700个基因是有表达,此时Max size为500,则该基因集不会纳入后续分析。基因集的基因数量过低或过高都会造成结果的假阳性、假阴性,一般的GO和KEGG在做超几何分析时候,也会适当限制。 Advanced Fields 1)Number ofmarkers:蝴蝶图butterfly plot中的markergenes的显示数量(但results结果中并没有看到官网所展示的蝴蝶图,因此可忽略);2)Plot graphs forthe top sets of each phenotype:每个表型phenotype需要绘制多少个基因集的GSEA plot,默认top 20,其它不绘制,后续可自行根据显著性结果的数量来适当调高参数,以保证所有显著性结果都有图片产生;3)Seed forpermutation:随机种子,由于分析过程中会随机置换,因此想让每次结果一致,这里需要设置同样的一个整数。 #如果想重现某次的结果,只是为了调整plot的产出数量,那么除参数必须一致以外,还需要获得上次分析的随机种子数值填写在该位置(可在上次分析result report报告结尾获得该信息); #其它参数一般不需要改,如果要自行改动,最好对GSEA的算法有一定理解,结合官网说明,进行适当修改。 03 运行与结果产出 参数设置完成后,点击下方Run后,左下的GSEA reports就显示该次分析的状态,并开始运行了。如果运行错误,会报错,显示error,可点击查看原因,如果运行完成,显示Success,则可点击Success,查看网页版结果报告。同时,也可点击下方的Show results folder可查看文件结果。 4、GSEA结果展示 打开GSEA结果文件夹后,结果文件非常多,GSEA并没有对结果进行很好的分类,导致文件看起来很杂乱。其实GSEA结果主要可分为三个部分,1)富集分析的结果tsv文件(制表符分隔文档),记录了每条基因集的富集分析结果,包括ES、NES、P值、FDR值等,区分表型phenotype中的Exp和Con后进行记录;2)基因集的GSEA plot图片(enplot,即enrichment plot);3)基因集对应的基因热图heatmap。还有一些关于每个基因集的网页版结果形式,用于展现上述结果,可自行打开查看即可。 01 网页报告 可打开GSEA结果文件夹中index.html网页版结果报告,也可点击GSEA软件界面的Success,查看网页版结果报告,网页版报告对结果可进行快速的全方位查看,同时方便跳转数据,其展示如下: 网页报告的信息总结如下:Enrichment in phenotype: Exp (3 samples)174个基因集中的12个基因集在表型Exp中上调;(表示12个基因集富集在Exp中,但是否可认为该基因集所代表的功能就出现上调或正向调节,仍需具体查看相关基因的作用导致的功能变化才能确定)2个基因集的FDR  第一部分,所有基因排序后的分布,横坐标代表所有基因的位置,数值总大小与表达矩阵的基因数量一样,纵坐标代表该基因的表达情况与表型的关联程度,从大到小排列,绝对值越大代表关联越强(可简单理解为类似差异倍数)。GSEA会根据基因表达情况与表型(分组)的关联程度进行排序(排序方式为信噪比Signal2noise,参数可设置),靠近横坐标0的基因代表与表型呈现正相关(Exp,positivelycorrelated),靠近横坐标30000的为负相关基因(Con,negatively correlated)。不同的颜色表示基因与表型的关联程度,红色表示基因在Exp中高表达,蓝色表示基因在Con中高表达;第二部分,用线条标记了该基因集中的基因出现在基因排序列表中的位置,黑线代表当前分析的基因集中的基因在所有基因排序中的位置;第三部分,绿色曲线为富集打分ES的动态过程,而遍历排序的基因列表是计算ES值的过程。ES值初始为0,然后顺着基因排序位置0处往后进行移动,当有排序基因中出现在该基因集中时则ES加分,反之不出现则减分,加减分值由基因与表型的相关性决定,最终完成全部排序基因的打分流程,形成完成的绿色曲线,而ES值最高(>0)或最低(0 |
【本文地址】