| 实用有机合成化学 | 您所在的位置:网站首页 › 吡啶高温分解 › 实用有机合成化学 |
实用有机合成化学
|
上周为大家简单介绍了邻位导向金属化(DoM)反应的发展历程,并从芳香羧酸及其衍生物作为底物开始讲起,展示了COOH、CONEt2等导向基团在苯环邻位C-H键官能化中的应用。 其中CONEt2具有很强的定位能力,即便底物中同时存在其他取代基,参与DoM反应时绝大多数情况下也都会选择性得到CONEt2邻位官能化的产物。还有人以空间位阻更大的CONiPr2作为导向基团,此时可使用更温和的碱nBuLi完成相应的转化。 噁唑啉 考虑到完成邻位官能化后将酰氨基水解重新转化为COOH一般需要在强酸、强碱等剧烈的条件下进行,有人则将芳香羧酸中的COOH转化为噁唑啉基团设计DoM反应。1975年,西巴-盖吉(Ciba-Geigy)公司的Heinz W. Gschwend博士与美国科罗拉多州立大学(Colorado State University)的Albert I. Meyers教授先后报道了使用4,4-二甲基-2-噁唑啉作为导向基团,完成了苯环邻位的选择性氘代。反应以nBuLi作为碱,在-45~-78 ℃的情况下便可高效进行,D2O还能换作CH3I、I2、tBuNCO等其他亲电试剂。构建4,4-二甲基-2-噁唑啉结构十分简单,一般从相应的芳香羧酸出发,在缩合剂的作用下与2-氨基-2-甲基丙醇缩合即可,参与DoM后噁唑啉基团在相对温和的酸性条件下便能水解为COOH。   4,4-二甲基-2-噁唑啉作为导向基团参与苯环的邻位官能化(图片来源:参考资料[3]) 4,4-二甲基-2-噁唑啉作为导向基团参与苯环的邻位官能化(图片来源:参考资料[3])法玛西亚-普强(Pharmacia and Upjohn)公司的Peter G. M. Wuts博士在合成包含呋喃并吡啶骨架的候选药物时需要以2-呋喃甲酸为原料,借助DoM的策略来实现。起初他将底物中的COOH转化为N-叔丁基氨甲酰基(CONHtBu),并以此为导向基团构建目标结构,却发现反应后很难将酰氨基水解,温度过高则会导致产物分解。于是,他想到将2-呋喃甲酸与N,N-二甲基肼缩合,进而设计DoM过程来解决这一问题。N,N-二甲基肼甲酰基(CONHNMe2)能在CuCl2、H5IO6等温和氧化剂的作用下氧化裂解重新得到COOH,无需苛刻的反应条件。这种导向基团同样可在苯环邻位C-H键的选择性官能化中发挥作用。   CONHNMe2作为导向基团参与苯环的DoM反应(图片来源:参考资料[6]) CONHNMe2作为导向基团参与苯环的DoM反应(图片来源:参考资料[6])4,4-二甲基-2-噁唑啉参与的DoM反应在医药研发领域也得到了推广应用。1981年,美国G. D. Searle公司的Charles R. Ellefson博士在合成苯并噻吩并四氢异喹啉类多巴胺拮抗剂时从二苯并噻吩-1-甲酸(4)出发,在SOCl2的作用下转化为对应的酰氯,随后与2-氨基-2-甲基丙醇缩合,形成噁唑啉取代的二苯并噻吩5。5经nBuLi邻位去质子化后与环氧乙烷反应,得到苯乙醇中间体6。后者在酸性条件下回流加热,促使噁唑啉基团水解并进一步发生分子内缩合环化,由此构建内酯结构7,7再通过后续转化得到目标产物。  苯并噻吩并四氢异喹啉类多巴胺拮抗剂的合成(图片来源:参考资料[7]) 苯并噻吩并四氢异喹啉类多巴胺拮抗剂的合成(图片来源:参考资料[7])氰基 氰基(CN)作为导向基团同样很少使用nBuLi作为碱,主要在于DoM过程中后者可直接对CN亲核加成。有人还尝试使用空间位阻较大的LDA对其邻位去质子化,体系温度较低时攫氢效率不理想,升温则不可避免地伴随着一定量的N,N-二异丙基苯甲酰胺副产物,即部分LDA对CN亲核进攻。 1983年,美国伊利诺伊大学厄巴纳-香槟分校(University of Illinois at Urbana-Champaign)的J. C. Martin教授提出了一种全新的理念来设计DoM反应——原位捕获(in situtrapping)。相比于上文提及的nBuLi、sBuLi等烷基锂试剂,这种方法一般利用空间位阻更大的碱(如LiTMP)来完成苯环的邻位导向锂化,但其碱性较弱,只能将苯环的邻位部分去质子化,形成的锂化中间体也相对不稳定。不过,由于体系中同时加入了能与LiTMP兼容的亲电试剂,一旦产生相应的锂化物种,后者便进一步与之反应,由此不断推动邻位官能化过程进行,实现目标转化。 基于以上策略,Martin教授以LiTMP作为碱、TMSCl作为亲电试剂,将两者混于THF溶液中,并在-78 ℃的条件下加入苯甲腈。体系缓慢恢复至室温,反应最终能以良好的收率得到苯环邻位硅烷化的产物。改变三种组分的用量还可调控邻位单、双取代产物的比例。当然,调整投入物料的顺序,预先将苯甲腈与TMSCl混合,后续加入LiTMP反应效果大体相同。  利用原位捕获策略设计CN作为导向基团的邻位硅烷化反应(图片来源:参考资料[8]) 利用原位捕获策略设计CN作为导向基团的邻位硅烷化反应(图片来源:参考资料[8])所谓亲电试剂与LiTMP兼容,从反应机制来看是由于两者的反应速率远小于苯环邻位去质子化的速率,因而也意味着亲电试剂的适用范围会受到一定的限制。后续人们还借助这种碱完成了B(OiPr)3对苯甲腈的邻位硼酸酯化。 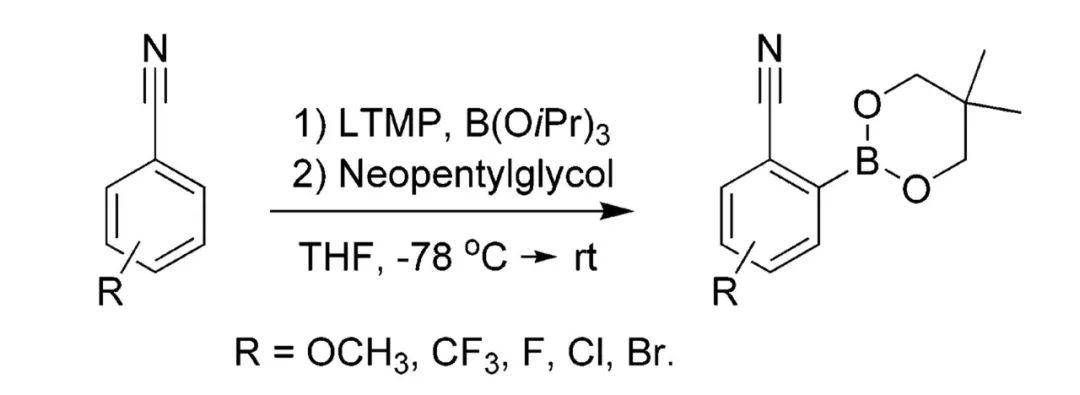 CN作为导向基团参与的邻位硼酸酯化(图片来源:参考资料[9]) CN作为导向基团参与的邻位硼酸酯化(图片来源:参考资料[9])2014年,德国慕尼黑大学(Ludwig-Maximilians-Universität München)的Paul Knochel教授还在此基础上发展了原位捕获转金属化过程。以2,4-二氯苯甲腈参与反应为例,他首先将这种底物与ZnCl2·2LiCl混合,并在-78 ℃的低温下加入LiTMP,5分钟内便可完成DoM/转金属化形成相应的苯基锌活性物种,随后在I2的作用下得到6位碘化的动力学产物。假使直接使用中等强度碱性的TMPZnCl·LiCl对2,4-二氯苯甲腈去质子化,反应发生在苯环H原子酸性最强的3位,最终得到热力学产物。ZnCl2·2LiCl还可换作MgCl2·2LiCl、CuCN·2LiCl等其他金属盐,结合更多不同类型的亲电试剂(如ArSSO2Ph(Ar =p-FC6H4)、3-溴环己烯)实现CN邻位的选择性官能化,进一步拓展了这种导向基团在有机合成中的应用。  原位捕获转金属化策略实现CN的邻位官能化(图片来源:参考资料[10]) 原位捕获转金属化策略实现CN的邻位官能化(图片来源:参考资料[10])酯基 Martin教授还利用原位捕获的策略考察了异丙氧甲酰基(COOiPr)作为导向基团参与苯环邻位C-H键硅烷化的反应情况,其间使用LiTMP作为碱,TMSCl作为亲电试剂,能以优异的产率得到目标产物。相比之下,苯甲酸乙酯的空间位阻较小,在相同的反应条件下更容易发生自缩合,邻位官能化的效率不够理想。 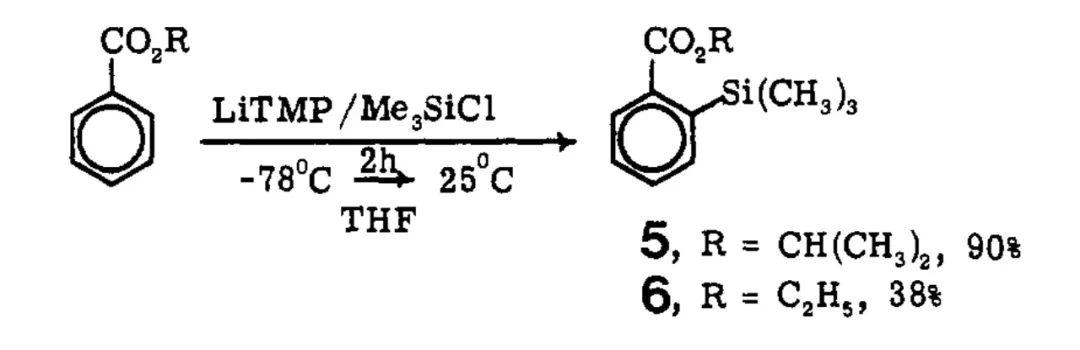 酯基作为导向集团参与苯环的邻位官能化(图片来源:参考资料[8]) 酯基作为导向集团参与苯环的邻位官能化(图片来源:参考资料[8])Knochel教授开发的原位捕获转金属化体系同样适用于酯基参与的DoM反应。此时选择乙氧甲酰基(COOEt)作为导向基团,3-氰基苯甲酸乙酯可以在低温下得到6位官能化的产物;换用TMPMgCl·LiCl作为碱,室温下反应则发生在2位。 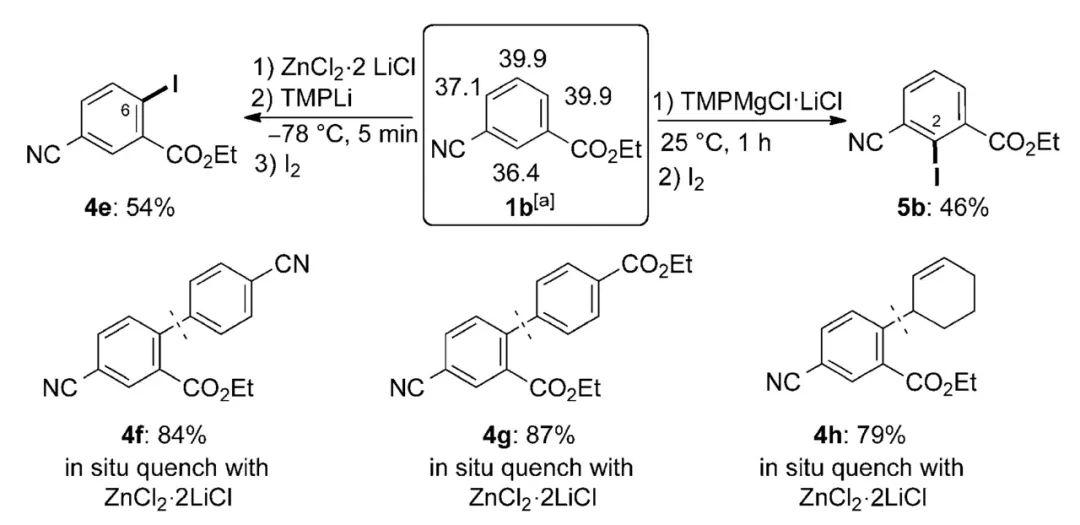 原位捕获转金属化策略实现COOEt的邻位官能化(图片来源:参考资料[10]) 原位捕获转金属化策略实现COOEt的邻位官能化(图片来源:参考资料[10])此外,他还分别利用(TMP)2Mg·2LiCl、(TMP)2Zn·2MgCl2·2LiCl作为碱设计COOEt的邻位官能化过程。后者不仅具有良好的DoM活性,还能与硝基(NO2)、甲酰基(CHO)等易接受亲核进攻的基团兼容,反应在室温条件下便可高效进行,适用性更加广泛。  (TMP)2Mg·2LiCl、(TMP)2Zn·2MgCl2·2LiCl作为碱参与COOEt的DoM反应(图片来源:参考资料[11][12]) (TMP)2Mg·2LiCl、(TMP)2Zn·2MgCl2·2LiCl作为碱参与COOEt的DoM反应(图片来源:参考资料[11][12])2016年,日本东京大学(the University of Tokyo)的Masanobu Uchiyama教授又报道了(TMP)2Cu(CN)Li2作为碱参与的邻位导向氨化、羟化反应,其中过氧叔丁醇(TBHP)用作羟化来源,苄氧基胺(BnONH2)为胺化试剂。这种方法适用于多种羧基衍生的导向基团,除了COOEt、CN,还包括此前讨论过的CONEt2、CONiPr2,甚至是其他类型的结构。  (TMP)2Cu(CN)Li2作为碱参与苯环的邻位C-H键羟化(图片来源:参考资料[13]) (TMP)2Cu(CN)Li2作为碱参与苯环的邻位C-H键羟化(图片来源:参考资料[13])从反应机制来看,芳香烃底物在这种碱的作用下首先形成相应的苯基铜中间体,但并非直接通过亲电官能化的途径得到最终产物。后续需经历配体交换、氧化加成、还原消除等步骤方能完成转化。  (TMP)2Cu(CN)Li2参考DoM可能的反应机制(图片来源:参考资料[13]) (TMP)2Cu(CN)Li2参考DoM可能的反应机制(图片来源:参考资料[13])以上内容便是COOH及其衍生结构应用于DoM反应的发展情况,后续我们还将为大家展示更多其他类型的导向基团在有机合成研究中的出色表现,敬请期待! 参考资料[1] Victor Snieckus, Directed ortho metalation. Tertiary amide andO-carbamate directors in synthetic strategies for polysubstituted aromatics.Chem. Rev.1990,90, 879.[2] Frédéric R. Leroux, Jacques Mortier.Directed Metalation of Arenes with Organolithiums, Lithium Amides, and SuperbasesinArene Chemistry: Reaction Mechanisms and Methods for Aromatic Compounds(Ed.: Jacques Mortier), VCH, Weinheim,2015.[3] Heinz W. Gschwend et al., Ortho-lithiation of aryloxazolines.J. Org. Chem.1975,40, 2008.[4] Albert I. Meyers et al., Oxazolines. XVII. Regioselective metalation of 2-aryl oxazolines. Route to polydeuteriobenzoic acids.J. Org. Chem.1975,40, 3158.[5] Michael Reuman et al., The synthetic utility of oxazolines in aromatic substitution.Tetrahedron1985,41, 837.[6] Scott A. Pratt et al., Dialkylhydrazides for directed orthometalations.Tetrahedron Lett.2000,41, 3559.[7] Charles R. Ellefson et al., Synthesis and evaluation of 1,2,3,4-tetrahydro[1]benzothieno[2,3-h]isoquinolines as dopamine antagonists.J. Med. Chem.1981,24, 1107.[8] Timothy D. Krizan et al., In situ trapping of ortho-lithiated benzenes containing electrophilic directing groups.J. Am. Chem. Soc.1983,105, 6155.[9] Morten Lysén et al., Synthesis of Substituted 2-Cyanoarylboronic Esters.J. Org. Chem.2006,71, 2518.[10] Annette Frischmuth et al., New In Situ Trapping Metalations of Functionalized Arenes and Heteroarenes with TMPLi in the Presence ofZnCl2and Other Metal Salts.Angew. Chem Int. Ed.2014,53, 7928.[11] Christoph J. Rohbogner et al., Magnesiation of Weakly Activated Arenes UsingTMP2Mg·2LiCl: Synthesis oftert-Butyl Ethyl Phthalate.Org. Synth.2009,86, 374.[12] Stefan H. Wunderlich et al.,(TMP)2Zn·2MgCl2·2LiCl: A Chemoselective Base for the Directed Zincation of Sensitive Arenes and Heteroarenes.Angew. Chem. Int. Ed.2007,46, 7685.[13] Noriyuki Tezuka et al., Direct Hydroxylation and Amination of Arenes via Deprotonative Cupration.J. Am. Chem. Soc.2016,138, 9166.⤵️喜欢我们的内容,欢迎关注@药明康德市场部!或者点赞、评论、分享给其他读者吧! |
【本文地址】