| Neuron | 您所在的位置:网站首页 › 可塑性和可固性一样吗 › Neuron |
Neuron
|
供稿 | abby 西安交通大学 排版 | AiBrain 编辑团队 我们每天和很多人打交道,不仅要结识新面孔,也要记住老面孔,这个过程就非常依赖于我们的“社交记忆”。社交记忆是指动物识别同物种其他动物的能力,是社会行为中非常重要的部分。海马是情景记忆形成的关键结构,在精神分裂症和躁郁症患者中,社交记忆缺失可能与海马体的异常有关,其中海马体的CA2区域尤为特别。 研究表明,海马CA2区是社交记忆形成的关键区域,更好地理解CA2的功能可能有助于了解以及治疗以社会行为改变为特征的疾病,如自闭症、精神分裂症和双相情感障碍等。 科学家们长期以来认为,海马在我们记忆日常生活中某人、某事、某地和某个时间的能力中起至关重要的作用。研究表明,海马的不同亚区具有不同的功能。例如,齿状回在区分相似环境中发挥重要功能,而CA3使得我们能够记起来自不完整线索的记忆,CA1区对于所有形式的记忆形成均至关重要。而CA2区是社交记忆形成的关键区域,在社交互动过程中,增加CA2区血管加压素的释放大大延长了社交记忆的持续时间。 CA2区的抑制性传递的可塑性可能有助于记忆的形成,了解海马CA2区域的功能是治疗和改善精神疾病或神经退行性疾病中观察到的社交记忆损伤的最佳途径。 北京时间2022年7月19日,巴黎西岱大学Vivien Chevaleyre团队在Neuron期刊在线发表题为“Sequential inhibitory plasticities in hippocampal area CA2 and social memory formation”的研究论文,揭示了海马CA2区序列抑制可塑性与社交记忆形成的神经机制。  海马CA2区中间神经元(PV+ INs)的抑制性传递受到了一种由δ阿片受体(DOR)激活介导的独特的长期抑制(iLTD),这种可塑性在青春期与社交记忆中同时出现。此外,药物阻断DOR或下调DOR表达可降低社交记忆。由此表明DOR可塑性受损可能是精神障碍中社交记忆缺陷的基础。 在这项研究中,作者证实了DOR可塑性可由大麻素1型受体(CB1R)介导的另一种抑制性可塑性进行诱导。首先,作者假设在CA2区诱导DOR-iLTD后,PV-CA2-PC突触的抑制可能允许次级突触可塑性的表达。实验发现,使用DOR激动剂孵育海马脑片20分钟后,电刺激CA3所引起CA2中的场突触后电位(fPSP)振幅持续增加(图1A)。该结果表明,DOR-iLTD允许另一种可塑性。接下来,作者验证了这种可塑性是否会在100 Hz电刺激后出现。 通常情况下,CA3兴奋性输入在CA2区不表达长时程增强(LTP),DOR激活引起的fEPSP幅度增加完全取决于DOR-iLTD。用100 Hz电刺激诱导DOR-iLTD导致fPSP振幅长期增加(图1B)。接下来,作者研究了fPSP振幅的第二次增加是由于兴奋性传递的LTP还是由抑制性突触的进一步LTD引起的。上述结果证实,在DOR介导的可塑性存在的情况下,允许诱导需要激活CB1R的次级抑制可塑性。 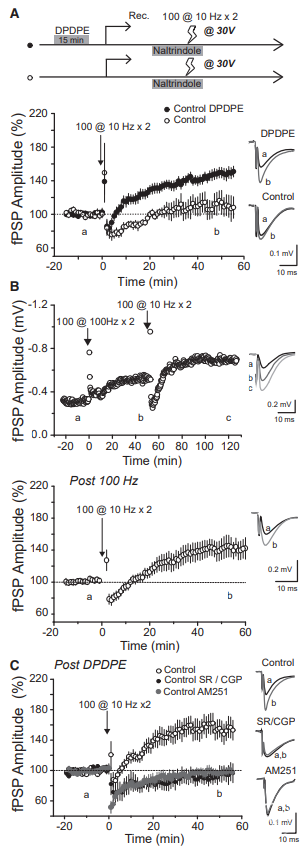 图1 在DOR介导的可塑性存在的情况下,CB1R介导的可塑性 如果激活CB1R会调节CA2区的抑制性传递,那么应用CB1R激动剂应导致fPSP振幅增加。作者发现短暂应用CB1R激动剂ACEA可导致fPSP幅度持续增加(图2A)。这种增加完全是通过抑制性传递的减少导致的,因为GABAR阻滞剂可以阻止这种增加(图2A)。 接下来,通过全细胞电压钳记录监测在AMPA/NMDA受体阻滞剂(D-AP5和NBQX)存在下+10 mV的抑制性突触后电位(IPSC)。结果证实应用ACEA导致IPSC振幅持续下降(图2B)。在海马中,CB1R主要在GABA能神经元末端表达,在那里它们起到减少神经递质释放的作用。因此,ACEA应用导致两个连续IPSC的配对脉冲比(PPR)显著增加,该测量值与释放概率呈负相关(图2B)。 此外,ACEA应用也导致自发IPSC频率降低(图2C),但没有改变其振幅。这些数据表明激活CB1R导致GABA释放概率降低。 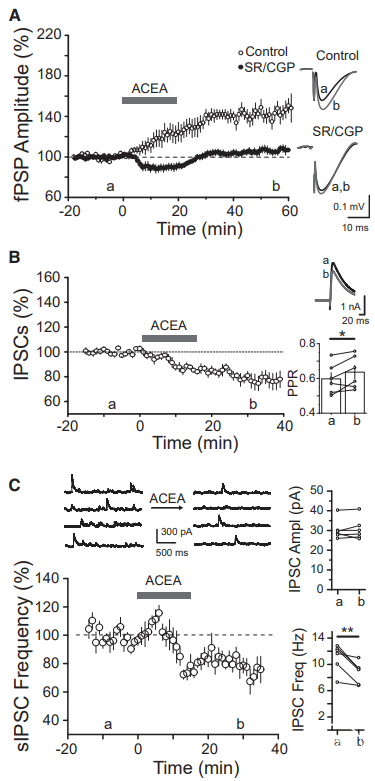 图2 激活CB1Rs可减少GABA释放和抑制CA3-CA2兴奋性传递 接下来,作者研究了CB1R介导的CA2区可塑性的潜在细胞机制。实验发现,虽然30V下的10 Hz刺激方案能够有效触发fPSP幅度的持续增加(图3A),但10V下的相同刺激几乎没有作用(图3A)。此外,与可以用100 Hz刺激诱导的DOR-iLTD相比,在DPDPE培养后,100 Hz刺激未能诱导fPSP幅度的任何显著增加。对这些发现的一种可能解释是,CB1R可塑性可能需要CA2-PNs激发动作电位(APs)进行诱导。 事实上,在基础条件下,CA3输入不会引起CA2 PNs中的APs,但在DOR-iLTD后会引起APs。因此,作者使用PNs的全细胞记录来测试CA2-PNs激发AP是否足以引起这种可塑性。实验结果证实EPSP-IPSP中去极化成分大幅度持续增加(图3B)。这种增加取决于抑制性传输,因为它可以被GABAR阻滞剂阻断(图3B)。它还依赖于CB1R的激活,因为它可以被AM251阻止(图3B)。 这些实验是在没有预先诱导DOR-iLTD的情况下进行,证明AP单独激发足以诱导CB1R可塑性。然后,作者分析了这期间诱发的AP数量与可塑性百分比。实验发现,30-60个Aps可引起PSP幅度显著增加(图3C)。为了直接测量抑制性传输的变化,作者记录了-70 mV时的IPSC幅度。为了诱导CB1R-iLTD,研究人员在诱导期间瞬时切换到电流钳以诱发APs。结果发现,在PN中10 Hz AP激发可引起IPSC振幅的LTD(图3D)。此外,iLTD诱导导致两个连续IPSC的PPR显著增加(图3D)。这些实验表明,在CA2 PNs中激活AP足以诱导CB1R-iLTD。CB1R可塑性可能依赖于DOR-iLTD在CA3输入刺激下激活PNs AP放电的能力。作者使用CACNG5-cre小鼠系在CA2 PNs中表达抑制性恐惧。在DPDPE培养的切片中,应用氯氮平N-氧化物(CNO)后10 Hz刺激下CA3的输入不会诱导任何可塑性(图3E和3F), 而未应用CNO的切片中10 Hz刺激下表达了DREADDS。这些结果表明,在CA2 PNs中激活AP是必要的,并且足以引起CB1R-iLTD。 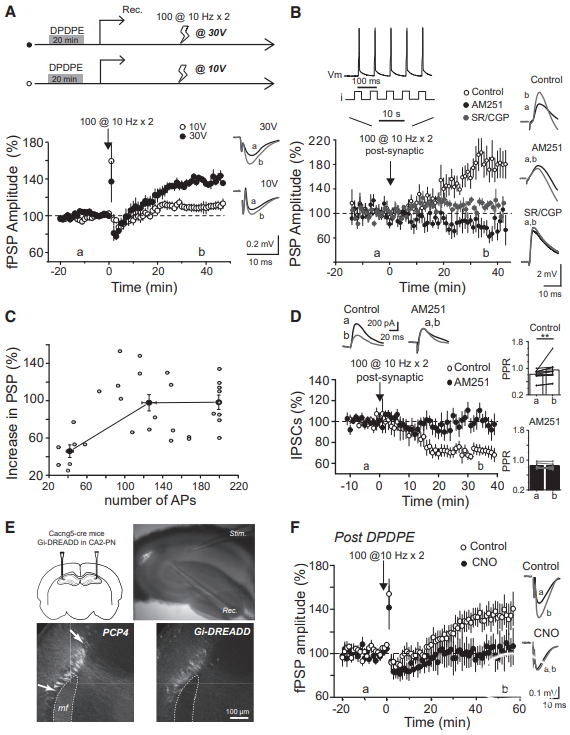 图3 CA2中PN AP放电足以诱导CB1R介导的可塑性 在CA1区,在SC刺激后,通过激活I组代谢型谷氨酸受体(mGluR-Is)可诱导CB1R-iLTD,该受体触发内源性大麻素(eCB)2-花生四烯酸甘油(2-AG)的释放,独立于细胞内Ca2+。 为了确定CA2 PNs中是否存在类似机制,作者观察了mGluR-I激动剂DHPG对fPSP的影响。当GABARs被阻断时,DHPG诱发fEPSP大而持久的抑制(图4A)。当DHPG应用于抑制传递完整的切片时,fPSP的抑制较小(图4A),表明DHPG也可能降低抑制传递。因此,作者在+10 mV时直接监测IPSC来测试DHPG的效果。DHPG应用引起IPSC振幅的瞬时降低,随后出现较小但持久的抑制(图4B)。在AM251存在的情况下,DHPG应用引起了类似的IPSC抑制(图4B)。在这两种情况下,DHPG诱导PPR无显著增加。 这些数据表明,mGluR-Is激活减少了CA2区的抑制性传递,但和CB1R无关。接下来,研究人员验证了在CA2 PNs中螯合Ca2+后CB1R可塑性是否改变。在记录吸管中加入BAPTA后,通过突触后10 Hz刺激诱导的PSP幅度去抑制性增加被完全消除(图4C),而在没有BAPTA的记录中观察到PSP增加(图4C)。 因此,AP放电期间细胞内Ca2+的增加是CB1R-iLTD诱导的必要条件,同时可能导致eCB释放。同时,与交叉对照组相比,当二酰甘油脂肪酶抑制剂四氢脂抑素(2-AG)应用于切片时,10 Hz刺激不会引起fPSP振幅的任何持续增加(图4D)。因此,2-AG可能是CA2区可塑性期间释放的eCB。  图4 CA2区的eCB释放取决于Ca2+的增加和DAG-L,不依赖于mGluR-Is激活 为了研究CB1R-iLTD后CA2区AP放电的变化,作者进行了细胞外记录,以监测CA2锥体层的群体峰值(PSs)。在10 Hz电刺激后,PS振幅进一步增加(图5A)。 因此,通过诱导CB1R-iLTD,可以进一步增加PV+INs处DOR-iLTD诱导的AP放电。虽然通过对所有CA2 PNs的全细胞记录观察到DOR-iLTD,但并非所有CA2 PNs都能够在DOR可塑性后触发APs。因此,作者假设只有在DOR可塑性后激发APs的细胞才会表达CB1R可塑性。为了验证这个问题,作者进行了细胞连接记录,以检测对CA3输入刺激的AP放电。 首先通过应用DPDPE诱导DOR可塑性,并测量AP激发概率。正如所料,在应用DPDPE后,只有一小部分小区触发了AP(图5B)。然后,对CA3输入施加10 Hz的刺激,并再次测量AP激发概率。与作者的预测一致,DPDPE后未触发APs的细胞不太可能在10 Hz刺激后触发APs(图5B)。然而,在DOR激活后激发AP的细胞在10 Hz刺激后表现出AP激发概率的显著增加(图5B)。 因为实验中在DPDPE应用后触发了一小部分CA2-PNs的AP,那么这些细胞的激发是否具有优先。 为了回答这个问题,作者用DPDPE诱发DOR可塑性,然后进行细胞贴附记录。有趣的是,在DOR可塑性后激发APs的绝大多数PNs位于表层,靠近SR,而在深层(接近SO)的28个PNs中只有1个激发APs(图5C和5D)。虽然深PNs在DPDPE后不会触发AP以响应两次CA3输入刺激,但它们仍有可能在10 Hz刺激期间触发足够的AP以表达CB1R可塑性。 通过细胞连接记录,作者量化了10 Hz刺激期间诱发的AP数量。这种刺激在浅层细胞中诱发162.9±14.3 APs(图5E),但在深层细胞中仅诱发38.3±18.6 APs。因此,在10 Hz刺激后,深PNs可能很少或没有CB1R可塑性。 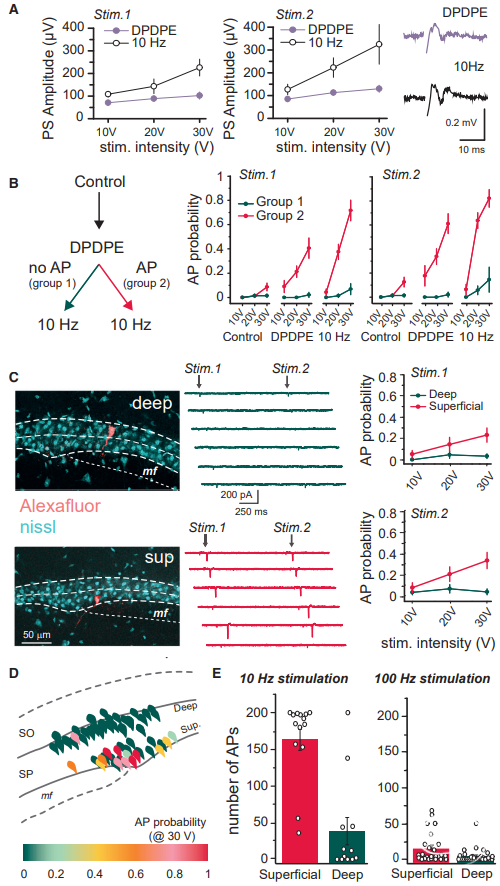 图5 CB1R-iLTD增加AP触发 先前的研究表明,DOR可塑性可能参与社交记忆的形成,这一过程依赖于CA2区。为了确定CB1R-iLTD是否也可能在社交记忆形成中发挥作用,作者使用了几种方法来证实。首先,在背部CA2区(图6A)双侧植入套管,并在五次社交记忆试验(图6B)之前加入AM251或空载体。在注射空载体的小鼠中,在实验2-4期间观察到与熟悉小鼠的相互作用时间显著减少(图6C和6D)。 相反,与试验1相比,注射AM251的小鼠在试验2-4中的相互作用时间没有减少(图6C和6D)。在AM251和空载体注射小鼠中,在第5次试验期间与新小鼠之间的相互作用水平与第一次与新小鼠之间的相互作用水平相似。第二天,对照组小鼠与现在熟悉的小鼠的相互作用时间缩短,表明社交记忆至少持续24小时。 有趣的是,前一天灌流AM251的小鼠在试验6期间的相互作用时间与试验1相似。然而,在试验6和7之间观察到交互作用显著减少,这表明这些小鼠在CA2区没有CB1R阻断,能够形成社交记忆。接下来,我们证实了AM251注射后的社交记忆缺陷是否是由于社交能力受损所致。实验发现,与对照组相比,空载体和AM251注射小鼠均对小鼠表现出强烈的偏好(图6E)。 这些数据表明,CB1Rs的激活在社交记忆形成中起作用,但在社交能力中不起作用。 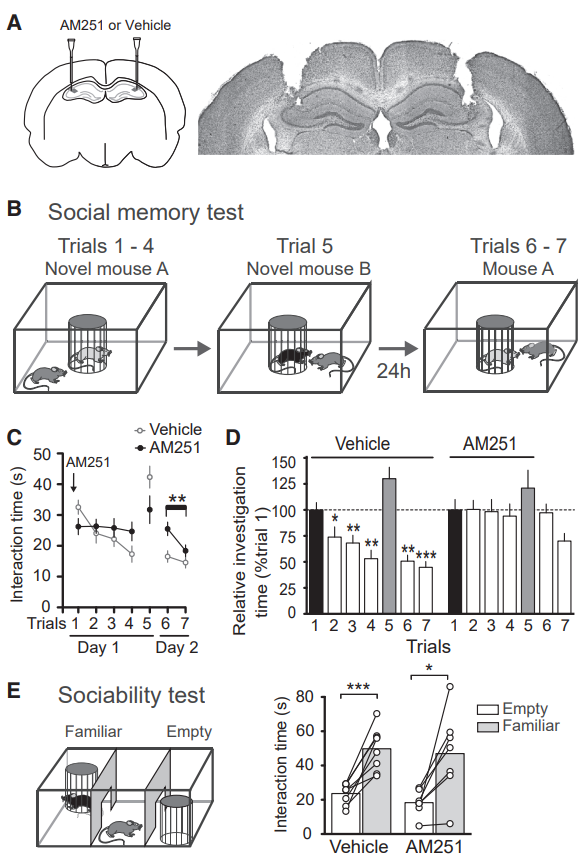 图6 在CA2区阻断CB1R可以在不改变社交能力的情况下阻止社交记忆 如果CB1R-iLTD是社交记忆形成所必需的,那么这种可塑性可能是在社会互动过程中诱导的。 作者像之前一样用社交记忆方案(试验1-4)处理小鼠,然后在5分钟后处死小鼠,并研究社会互动(S.I.)后的突触可塑性。对照组小鼠接受了4次相同环境的处理,没有社交互动(图7A)。 首先,作者发现S.I.小鼠(图7B)和对照小鼠中由100 Hz刺激诱导的DOR依赖性fPSP振幅增加减少,表明S.I后DOR可塑性部分被阻断。然后,作者检查了CB1R介导的可塑性是否也被S.I改变。实验发现,与对照小鼠相比,S.I.小鼠(图7C)由10 Hz刺激诱导的可塑性显著较小。为了探讨10 Hz介导可塑性的降低是否是由于eCB释放减少,作者还测试了ACEA的作用。 实验发现,在S.I.小鼠中,应用ACEA后fPSP幅度的增加显著降低(图7D)。然后,作者研究了S.I.是否减少了CB1R敏感性的抑制性传递。与对照组相比,ACEA对对照组小鼠IPSC幅度的影响(图7E),而在S.I.小鼠中显著降低。如果CB1R-iLTD是由社交记忆诱导的,作者预测CA2-PNs更有可能在CA3输入刺激时触发APs。事实上,与对照组小鼠切片相比,在S.I.小鼠切片中,CA3刺激导致大的PS(图7F)。切片中的结果证实了DOR和CB1R可塑性之间的顺序性。然而,这两种可塑性也有可能在体内同时诱发。 为了解决这个问题,作者与一只新的小鼠进行了一次5分钟的相互作用(5分钟S.I.),然后在5分钟内处死试验小鼠。100 Hz刺激诱导的DOR可塑性(图7G)约为对照小鼠诱发的幅度的一半,表明DOR-iLTD部分闭塞。相反,当提供10 Hz刺激以诱导CB1R可塑性时,在5分钟S.I.(图7H)和对照小鼠之间,fPSP的增加是相同的。 总之,这些数据有力地支持了CB1R可塑性在社交记忆形成中的作用,同时表明在社交记忆形成过程中DOR和CB1R可塑性的顺序性发生在体内。 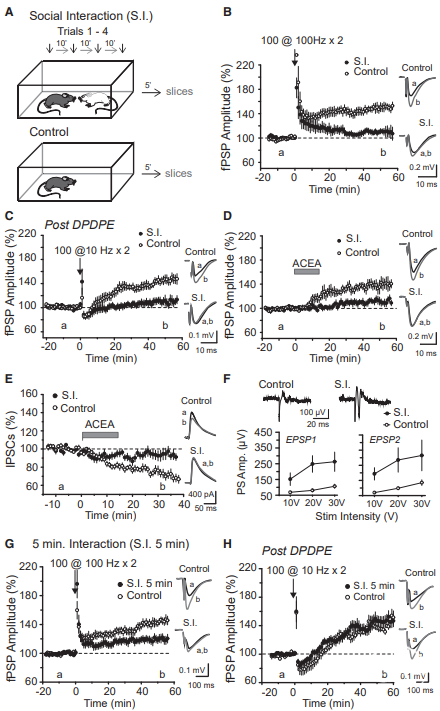 图7 社交记忆的形成阻碍了CB1R介导的可塑性 在22q11.2缺失综合征(DS)的小鼠模型中,社会识别记忆受损。这种损伤可能是由于这些小鼠的CA2-PNs更超极化,因此即使在DOR-iLTD后也会阻止AP放电。因此,作者验证了这些小鼠的CB1可塑性是否改变。在野生型(WT)同窝对照中,DPDPE孵育后的10 Hz刺激诱导fPSP大幅增加(图8A)。相比之下,10 Hz的刺激导致22q11.2DS小鼠(LgDel)切片中的fPSP振幅略有增加(图8A)。这种损伤可以解释为,在22q11.2DS小鼠中,CA2-PNs更为超极化,激发更少的AP,并且不释放eCB。 或者,CB1R介导的可塑性的分子途径可能受损。为了验证这一点,作者进行了全细胞记录,并使用了10 Hz去极化诱导AP在PN中激发。实验证实WT和LgDel小鼠之间的PSP振幅的去抑制增加是相同的(图8B)。 因此,LgDel小鼠CB1可塑性受损可能是由于AP放电受损,而不是CB1R激活下游信号受损所致。在22q11.2DS小鼠中,由于通过K+通道TREK-1的电导更高,CA2-PNs更超极化。减少CA2 PNs中的TREK-1电导可以恢复22q11.2DS小鼠的社交记忆。因此,作者假设阻断TREK-1通道可以恢复CB1R可塑性。用DPDPE诱导DOR可塑性,然后记录fPSP并应用氟西汀阻断TREK-1通道。结果证实,氟西汀的应用导致fPSP略有增加(图8C)。然后,在氟西汀应用结束时应用10 Hz的CA3输入刺激来诱导CB1R可塑性。结果fPSP幅度大幅增加(图8C),与WT小鼠诱发的幅度无明显差异。阻断TREK-1通道可能允许可塑性诱导,因为它允许PNs触发AP。 事实上,虽然LgDel小鼠DOR可塑性后几乎没有诱发PS,但氟西汀的应用足以揭示AP对CA3输入刺激的反应。总之,这些结果与CB1R可塑性在社交记忆形成中的作用是一致的。在社交记忆受损的小鼠模型中,不仅CB1R可塑性发生改变,还可以通过一种被证明可以挽救社交记忆的操作来挽救,即通过TREK-1阻断使CA2-PNs去极化。 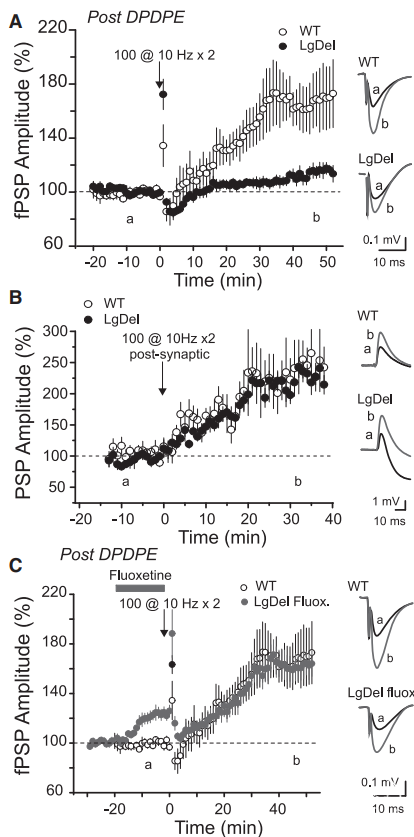 图8 在22q11.2 DS小鼠模型中,CB1可塑性受损 总而言之,该研究证实了一种由大麻素1型受体激活介导的新型抑制可塑性(C/B1R-iLTD)。令人惊讶的是,CB1R-iLTD需要预先诱导DOR-iLTD。CA2中CB1R的阻断完全阻止了社交记忆的形成。 此外,DOR和CB1R介导的可塑性在连续的社会互动中发生。最后,在社会认知受损的精神分裂症小鼠模型中,CB1R-iLTD发生改变,但通过一种也能挽救社交记忆的操作可以挽救。 这些数据有力地支持了CB1R可塑性在社交记忆形成中的作用,并介绍了如何使用有利于CA2区CB1R可塑性的潜在治疗来改善在精神疾病或神经退行性疾病中观察到的社交记忆损伤。 AiBrain内容团队为大家整理了文章的pdf,如有需要,请公众号后台留言“pdf”或扫码添加AiBrain助手微信获取。 参考文献: 【1】Loisy M, Bouisset G, Lopez S, Muller M, Spitsyn A, Duval J, Piskorowski RA, Verret L, Chevaleyre V. Sequential inhibitory plasticities in hippocampal area CA2 and social memory formation. Neuron. 2022 Jul 15:S0896-6273(22)00551-7. doi: 10.1016/j.neuron.2022.06.013. Epub ahead of print. PMID: 35858622.
 ✦往期精彩回顾✦

声明:脑医汇旗下神外资讯、神介资讯、脑医咨询、AiBrain所发表内容之知识产权为脑医汇及主办方、原作者等相关权利人所有。未经许可,禁止进行转载、摘编、复制、裁切、录制等。经许可授权使用,亦须注明来源。欢迎转发、分享。
|



【本文地址】
| 今日新闻 |
| 推荐新闻 |
| 专题文章 |