| 三代测序数据分析实战 | 您所在的位置:网站首页 › 三代测序基因检测 › 三代测序数据分析实战 |
三代测序数据分析实战
|
三代测序数据分析实战
主讲人:徐怡
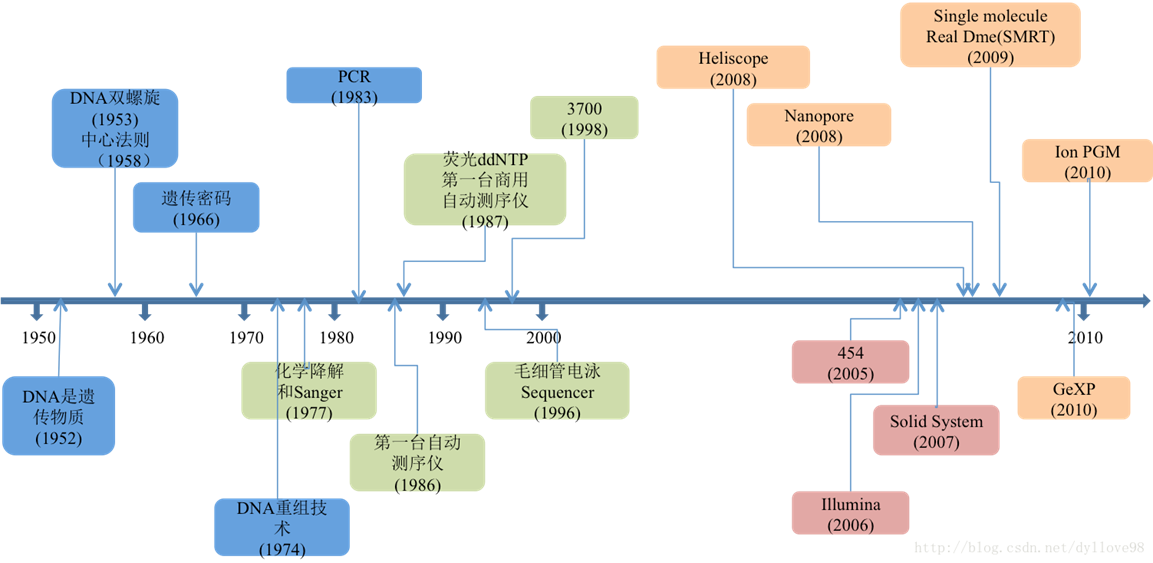
浙江大学医学院博士,迪安诊断研发中心生物信息工程师,负责 NGS 遗传检测。 背景介绍从1977年第一代DNA测序技术(Sanger法)发展至今三十多年时间,测序技术已取得了相当大的发展,从第一代到第三代乃至第四代,测序读长从长到短,再从短到长。虽然就当前形势看来第二代短读长测序技术在全球测序市场上仍然占有着绝对的优势位置,但第三和第四代测序技术也已在这一两年的时间中快速发展着。测序技术的每一次变革,也都对基因组研究,疾病医疗研究,药物研发,育种等领域产生巨大的推动作用。 第三代测序技术目前已经成为科研领域不可或缺的一种主流技术,广泛应用于基因组 Denovo、全长转录本检测、宏基因组、重测序和变异检测等多个方向,并且在染色体结构变异(SV)的检测中有着不可替代的优势。 第三代测序技术目前存在着错误率较高的瓶颈,生物信息学分析软件也不够丰富,但是未来随着准确度的提升、平行测序能力和酶活性等问题的解决,第三代测序技术是未来发展的重要技术趋势,实现大规模商业化将是大势所趋。 测序技术发展历程
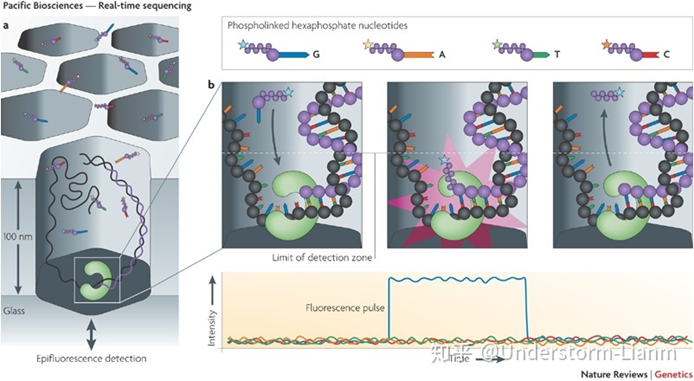
第三代测序技术是指单分子测序技术。DNA测序时,不需要经过PCR扩增,不仅实现了对每一条DNA分子的单独测序,并且避免了潜在的PCR扩增错误和偏好性。 第三代测序技术目前已经成为科研领域不可或缺的一种主流技术,广泛应用于基因组Denovo、全长转录本检测、宏基因组、重测序和变异检测等多个方向,并且在染色体结构变异(SV)的检测中有着不可替代的优势。 三代测序技术优点 读长长测序速度快三代测序设备小型化,便捷,稳定性好 三代测序技术缺点 成本偏高错误率较高生物信息学分析软件不够丰富 趋势未来随着第三代测序技术的准确度提升、平行测序能力和酶活性等问题的解决,第三代测序技术是未来发展的重要技术趋势,实现大规模商业化将是大势所趋。 三代测序平台及原理介绍 三大主流平台 测序方法/平台公司方法/酶平均测序长度技术原理HeliScopeHelicos边合成边测序/DNA聚合酶30-35 bp单分子荧光测序SMRTPacific Biosciences边合成边测序/DNA聚合酶100000 bp单分子荧光测序Nanopore sequencingOxford Nanopore电信号测序/核酸外切酶无限长纳米孔测序 SMRTPacBio SMRT(single molecule real time sequencing)技术应用了边合成边测序的思想,并以SMRT 芯片为测序载体。 基本原理: DNA 聚合酶和模板结合,4色荧光标记4种碱基(即dNTP),在碱基配对阶段,不同碱基的加入,会发出不同光,根据光的波长与峰值可判断进入的碱基类型。
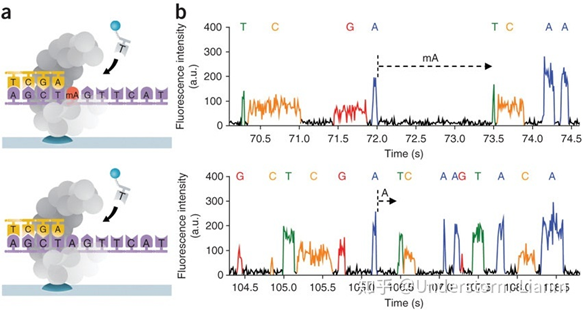
注:DNA 聚合酶是实现超长读长的关键之一,读长主要跟酶的活性保持有关,它主要受激光对其造成的损伤所影响。 测序步骤: (1)聚合酶捕获文库DNA序列,锚定在零模波导孔底部; (2)4种不同荧光标记的dNTP随机进入零模波导孔底部; (3)荧光dNTP被激光照射,发出荧光,检测荧光; (4)荧光dNTP与DNA模板的碱基匹配,在酶的作用下合成一个碱基; (5)统计荧光信号存在时间长短,区分匹配碱基与游离碱基,获得DNA序列; (6)酶反应过程中,一方面使链延伸,另一方面使dNTP上的荧光基团脱落; (7)聚合反应持续进行,测序同时持续进行。 优点: 可以通过检测相邻两个碱基之间的测序时间,来检测一些碱基修饰情况: 若碱基存在修饰,则通过聚合酶时的速度会减慢,相邻两峰之间的距离增大,可以通过这个来直接检测甲基化等信息。
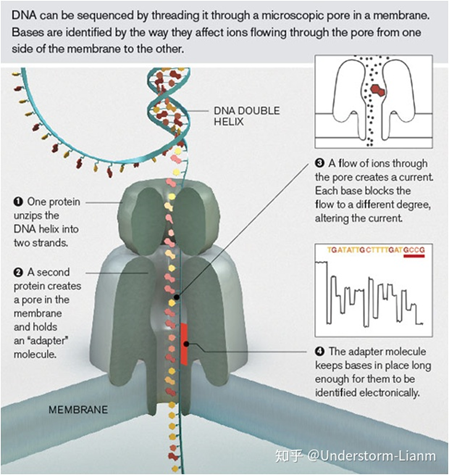
缺点: 测序错误率比较高,达到15%,这几乎是目前单分子测序技术的通病。 出错是随机的,因而可以通过多次测序来进行有效的纠错 Nanopore sequencing基本原理: 纳米孔测序设计了一种特殊的纳米孔,孔内共价结合有分子接头。当DNA 碱基通过纳米孔时,它们使电荷发生变化,从而短暂地影响流过纳米孔的电流强度(每种碱基所影响的电流变化幅度是不同的),灵敏的电子设备检测到这些变化从而鉴定所通过的碱基。
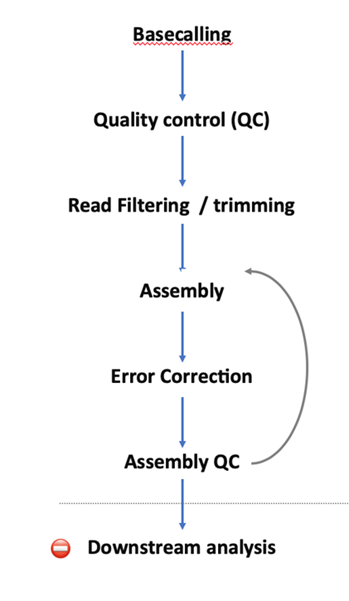
测序步骤: (1)解螺旋,将双链DNA解开成单链; (2)DNA单链分子通过一个孔道蛋白,孔道中有个充当转换器的蛋白分子; (3)DNA单分子停留在孔道中,有一些离子通过带来电流变化,而不同的碱基带来的电流变化是不同的; (4)转化器蛋白分子感受不同碱基的电流变化; (5)根据电流变化的频谱,应用模式识别算法得到碱基序列。 测序读长: 由于测序无需DNA聚合酶的链式反应,所以不存在DNA聚合酶的失活问题,理论上只要DNA分子不断开,就一直可以通过纳米孔,目前在对于人和大肠杆菌的测序中观测到的read是1Mb左右。 测序准确率: Nanopore测序准确率和Pacbio持平,为86%左右。而且起始位置正确率偏低,在大约100nt位置达到稳定,且错误为随机测序错误。 ==出错是随机的,因而可以通过多次测序来进行有效的纠错== 三代测序数据分析流程注:以下流程以假微型海链藻(Thalassiosira pseudonana) 基于 Nanopore sequencing 的基因组数据为例。
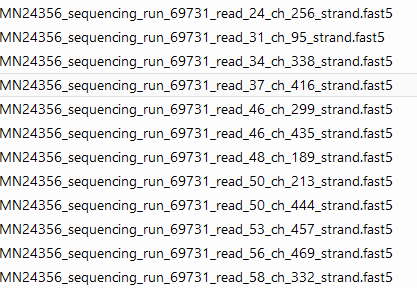
MinION 每个样本下机数据是一系列的fast5文件,如下:
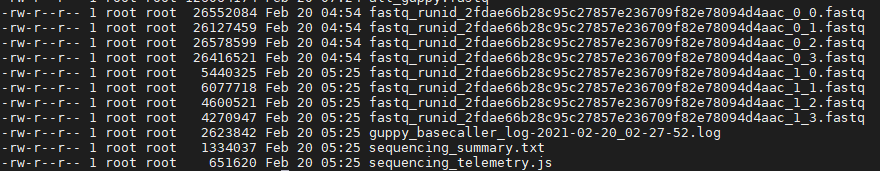
Guppy Guppy 是当前的“官方” ONT basecaller,基于basecalling的神经网络模型,将原始电子信号转换成碱基,生成fastq格式。除了basecalling之外,它还能进行低质量的reads 过滤、基于牛津纳米孔测序的adapter剪切等功能。 使用命令示例: guppy_basecaller –i /long-read-analysis/example_practice/0_rawdata –c /long-read-analysis/software/ont-guppy-cpu/data/dna_r9.4.1_450bps_hac –s /long-read-analysis/example_practice/1_basecalling --num_callers 4 --cpu_threads_per_caller 3 参数说明: –i 包含所有fast5文件的目录 -c 根据测序芯片和试剂盒的型号选择不同的配置文件,配置文件位于guppy的安装目录/data下 –s 结果输出目录 --num_callers basecalling 比较耗时,可将任务分拆同时进行 --cpu_threads_per_caller 每个任务分配的 CPU 线程生成结果如下:
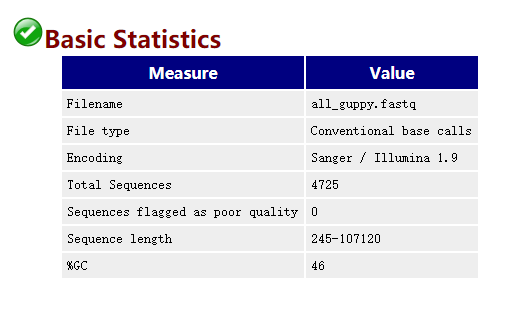
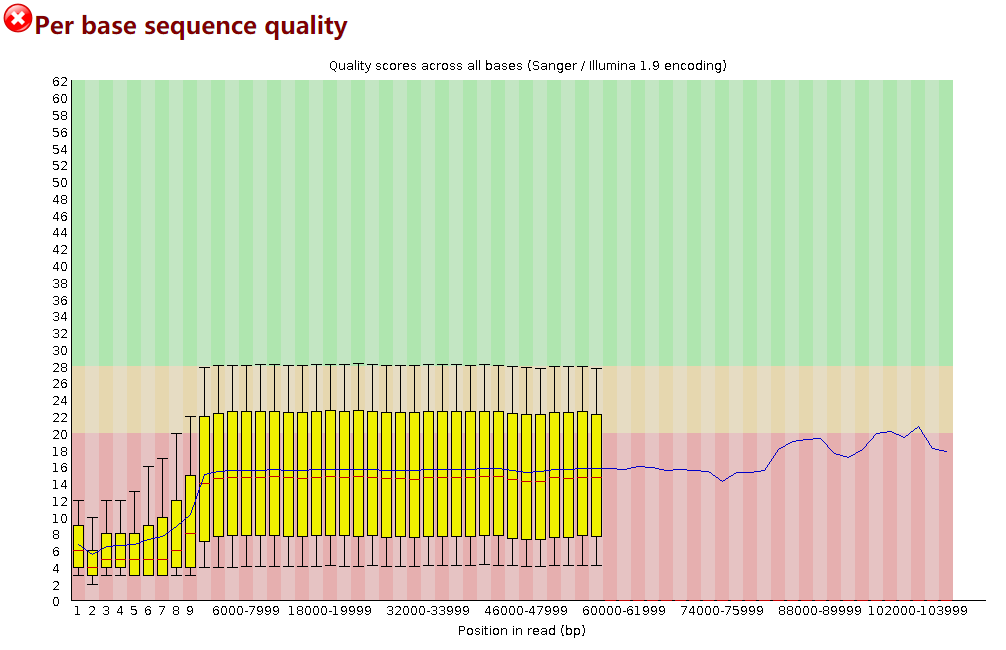
然后将所有fastq格式文件合并到一起: cat *.fastq > all_guppy.fastq 2. Quality Control of Raw Reads2.1 FastQC FastQC 是测序数据质控最常用的工具之一,支持Illumina、Oxford Nanopore和PacBio data 等各种平台。 使用命令示例: fastqc -o ./1_fastqc /long-read-analysis/example_practice/1_basecalling/all_guppy.fastq -t 4打开生成的结果文件all_guppy_fastqc.html:
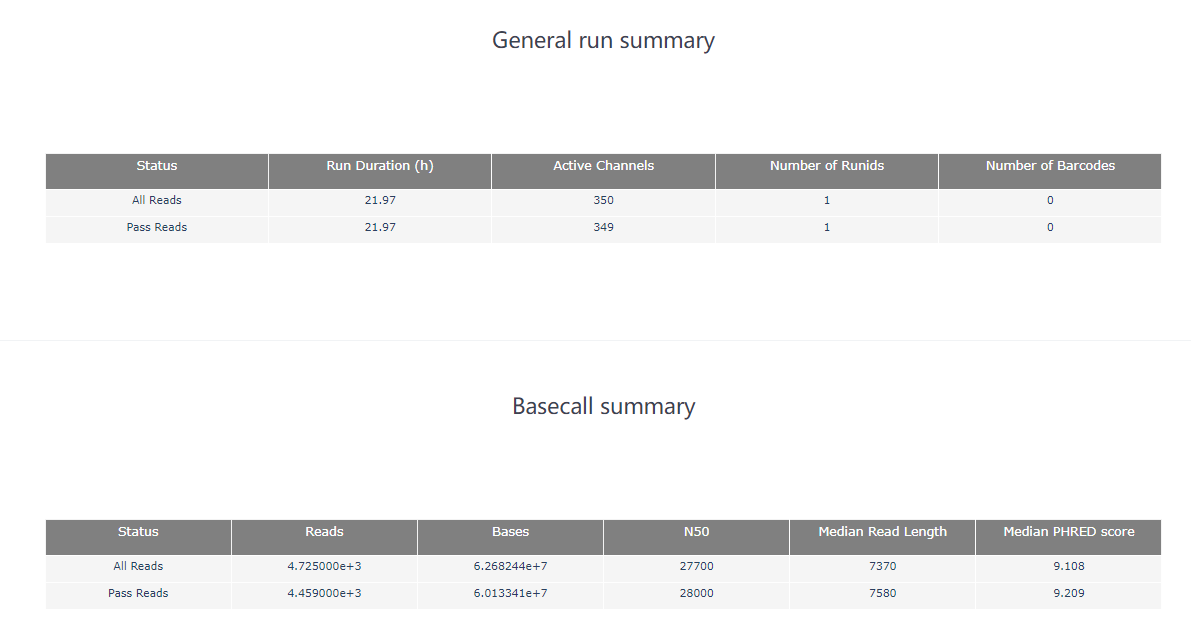
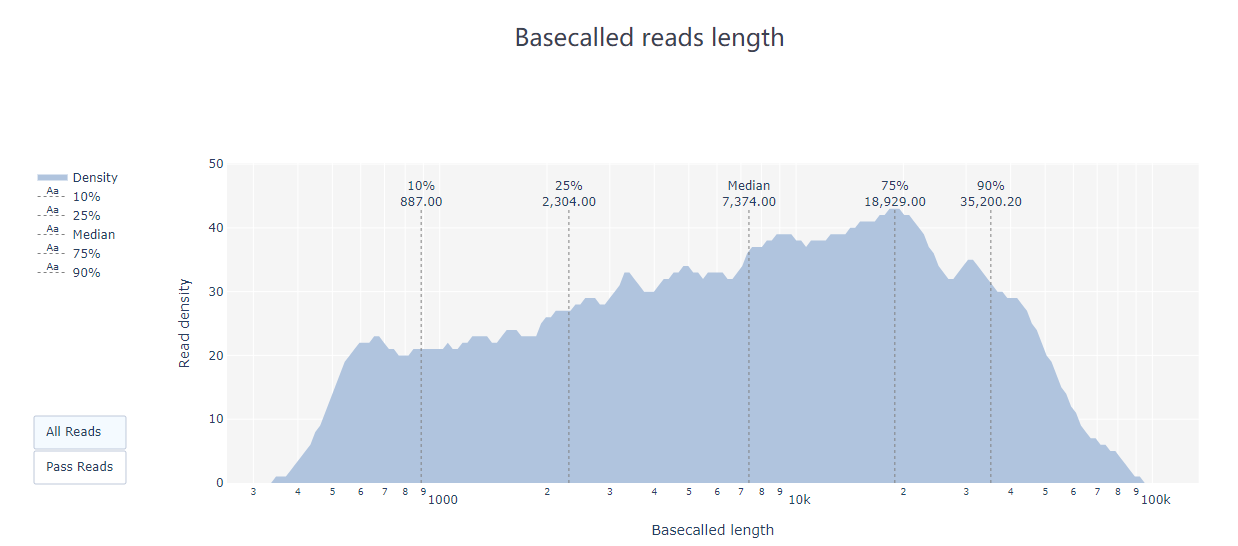
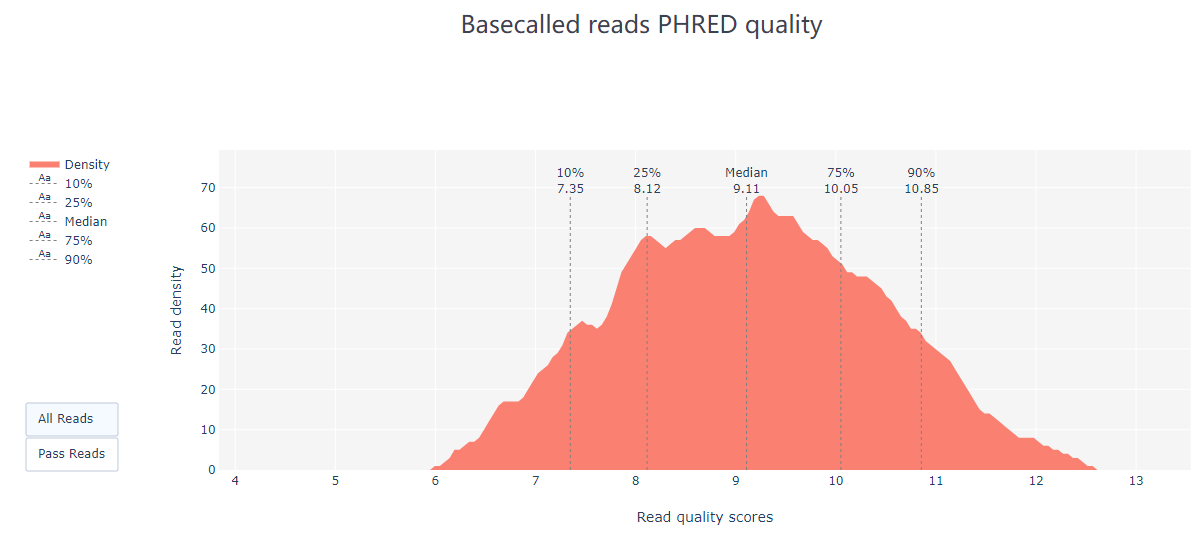
2.2 PycoQC PycoQC 是一种基于纳米孔数据的数据可视化和质控工具。与FastQC相比,它需要一个特定的sequencing_summary.txt 作为输入文件,该文件由Oxford nanopore basecaller (如Guppy或albacore basecaller)生成。 使用命令示例: pycoQC –f /long-read-analysis/example_practice/1_basecalling/sequencing_summary.txt –o ./2_pycoQC/all_guppy_pycoQC.html打开生成的结果文件all_guppy_pycoQC.html:
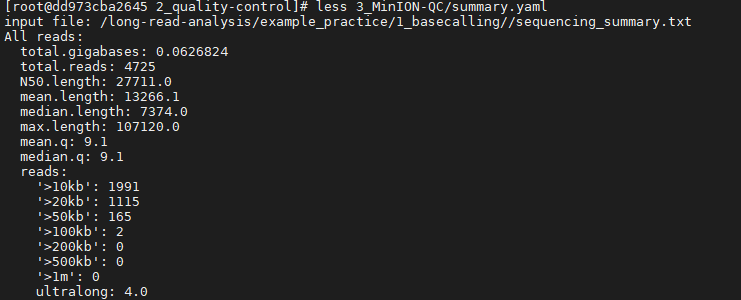
2.3 MinION_QC MinIONQC 也是一种基于纳米孔数据的数据可视化和质控工具,需要一个特定的sequencing_summary.txt 作为输入文件。与PycoQC相比,它能够比较多个测序结果的质控结果。 MinIONQC.R –i /long-read-analysis/example_practice/1_basecalling/sequencing_summary.txt -o ./3_MinION-QC查看结果文件summary.yaml:
Adapter Removal using PoreChop 使用命令示例: porechop –i /long-read-analysis/example_practice/1_basecalling/all_guppy.fastq -o ./porechopped.fastq --discard_middleRead trimming and filtering using NanoFilt 使用命令示例: NanoFilt -l 500 --headcrop 10 ./porechopped.fastq > ./nanofilt_trimmed.fastq 4. Genome Assembly由于第三代测序技术的高误差率,将PacBio和Oxford Nanopore等长读长数据组装成 contigs 对普通的第二代测序组装软件来说是一个挑战。在过去的几年里,越来越多的专门为长读长reads 设计的组装软件被发布,例如Canu、Flye、Shasta和miniasm。 不同的组装软件对不同的基因组有不同的作用。基因组大小、重复性、GC含量等因素都会影响组装软件的性能。最好的办法是运行多个组装软件,然后比较结果,经评估后决定使用哪一个软件的结果。 以下将介绍 minimap2-miniasm 的基因组组装流程: Genome Assembly with Minimap2 and Miniasm minimap2-miniasm 流程是组装长读长read的一种非常快速和高效的方法,个人比较推荐。 使用命令示例: #使用 minimap2 对序列进行自我比对,生成overlap校正文件 minimap2 –x ava-ont /long-read-analysis/example_practice/3_filtering-trimming/porechopped.fastq/nanofilt_trimmed.fastq /long-read-analysis/example_practice/3_filtering-trimming/porechopped.fastq/nanofilt_trimmed.fastq | gzip -1 > ./minimap.paf.gz #使用 miniasm 进行序列组装 miniasm -f /long-read-analysis/example_practice/3_filtering-trimming/nanofilt_trimmed.fastq ./minimap.paf.gz > miniasm.gfa #提取contigs序列保存为fasta格式 awk '/^S/{print ">"$2"n"$3}' miniasm.gfa > miniasm.fasta 5. Error correction5.1 Error Correction using Racon Racon 软件是为了补充minimap2/miniasm 流程而开发的,但可以用于任何长读长reads读取的组装结果。它提供了一个快速的一致性算法,可对二代短reads 和 三代长读长reads 进行校正。 使用命令示例: minimap2 /long-read-analysis/example_practice/4_genome-assembly/miniasm.fasta /long-read-analysis/example_practice/3_filtering-trimming/nanofilt_trimmed.fastq > ./minimap.racon.paf racon /long-read-analysis/example_practice/3_filtering-trimming/nanofilt_trimmed.fastq minimap.racon.paf /long-read-analysis/example_practice/4_genome-assembly/miniasm.fasta > ./miniasm.racon.consensus.fasta5.2 Error Correction using Minipolish 与 Racon 类似,Minipolish 是专门为校正 minimap2/miniasm流程的结果而编写。事实上,minipolish 是调用 Racon 来优化 miniasm 的结果,但与 Racon 不同的是,它读取和输出文件是miniasm 的GFA格式,而不是fasta 格式。 使用命令示例: minipolish -t 4 /long-read-analysis/example_practice/3_filtering-trimming/nanofilt_trimmed.fastq /long-read-analysis/example_practice/4_genome-assembly/miniasm.gfa > ./minipolished_assembly.gfa awk '/^S/{print ">"$2"n"$3}' minipolished_assembly.gfa > minipolished_assembly.fasta5.3 Pilon Pilon可以在Racon 之后运行,通过纠正插入/缺失(Indel)和单核苷酸多态性(SNPs) 的错误进一步提高组装质量。 Pilon使用命令示例: # index the consensus sequence bwa index /long-read-analysis/example_practice/5_error-correction/1_Racon/miniasm.racon.consensus.fasta # map reads bwa mem -t 5 /long-read-analysis/example_practice/5_error-correction/1_Racon/miniasm.racon.consensus.fasta -x ont2d /long-read-analysis/example_practice/1_basecalling/all_guppy.fastq > ./bwa_mapping.sam samtools view -Sb bwa_mapping.sam > bwa_mapping.bam samtools sort -o bwa_mapping.sorted.bam bwa_mapping.bam samtools index bwa_mapping.sorted.bam # run Pilon java -Xmx16G -jar /long-read-analysis/software/pilon/pilon-1.23.jar --genome /long-read-analysis/example_practice/5_error-correction/1_Racon/miniasm.racon.consensus.fasta --bam bwa_mapping.sorted.bam 6. Variant calling长读长测序的虽然可以检测单核苷酸多态性(SNPs),但是在检测长片段结构变异(SVs)方面更具优势。三代测序技术的较高错误率,使得对单核苷酸多态性的检测具有很高的挑战性。到目前为止,只有很少的工具能够对第三代测序数据进行 SNP 和 SV 的检测。 Sniffles Sniffles 主要用于检测长读长数据的SV,专门为 Pacbio 和 Oxford Nanopore数据设计,已经展现出了良好的性能。 使用命令示例: # map to genome minimap2 --MD -a /long-read-analysis/example_practice/Thalassiosira-pseudonana.chr17.fasta /long-read-analysis/example_practice/1_basecalling/all_guppy.fastq > mapped.sam # Convert to bam file samtools view -bS mapped.sam > mapped.bam # Sort the bam file samtools sort -o mapped.sorted.bam mapped.bam # create an index file samtools index mapped.sorted.bam # Run sniffles sniffles -m mapped.sorted.bam -v variants.vcf查看结果文件variants.vcf: 第三代测序技术展望第三代测序技术目前已经成为科研领域不可或缺的一种主流技术,目前已经在染色体结构变异(SV)的检测中有着不可替代的优势。 虽然第三代测序技术目前存在着错误率较高的瓶颈,生物信息学分析软件也不够丰富,但是未来随着准确度的提升、平行测序能力和酶活性等问题的解决,第三代测序技术是未来发展的重要技术趋势,实现大规模商业化将是大势所趋。 三代测序数据分析专题 内容:三代测序数据分析实战主讲人:徐怡时间:2021年3月06日 下午 2:00-5:00地点:线上 |
【本文地址】