| 靶向难成药靶点STAT的磷酸肽类抑制剂:花开两朵,各表一枝 STAT家族是信号转导和转录激活因子(signal transducer and activator of transc... | 您所在的位置:网站首页 › 一枝开两朵花叫什么 › 靶向难成药靶点STAT的磷酸肽类抑制剂:花开两朵,各表一枝 STAT家族是信号转导和转录激活因子(signal transducer and activator of transc... |
靶向难成药靶点STAT的磷酸肽类抑制剂:花开两朵,各表一枝 STAT家族是信号转导和转录激活因子(signal transducer and activator of transc...
|
来源:雪球App,作者: 医药魔方,(https://xueqiu.com/8965749698/260773541) STAT家族是信号转导和转录激活因子(signal transducer and activator of transcription)的缩写,它们是一类参与细胞内信号传导和基因表达调控的蛋白质。STAT家族包括STAT1、STAT2、STAT3、STAT4、STAT5a、STAT5b和STAT6,它们在哺乳动物中广泛存在,介导多种细胞因子和生长因子的信号通路。 点击下载:医药魔方-新药研发中的靶点演变 STAT3是STAT家族中最重要的成员之一,它在细胞的增殖、存活、分化、血管生成、免疫调节等多种生物学过程中发挥重要作用。STAT3的结构由6个功能保守结构域组成:氨基末端结构域(NTD)、螺旋卷曲结构域(CCD)、DNA结合结构域(DBD)、连接结构域(Linker)、SRC同源2结构域(SH2)和羧基末端反式激活结构域(TAD)。在正常细胞中,STAT3的激活是瞬时的,受到负调节因子的严格控制。当细胞受到刺激时,如白介素6(IL-6)、表皮生长因子(EGF)等,它们与相应的受体结合,导致受体上游的Janus激酶(JAK)或非受体酪氨酸激酶(Src)被磷酸化。然后,这些激酶将STAT3的酪氨酸残基705位点磷酸化,使其形成同源或异源二聚体,并转移到细胞核中,与特定的DNA序列结合,激活靶基因的转录。 然而,在多种人类癌症中,STAT3被持续或过度激活,通常与不良的临床预后有关。作为一个转录因子,STAT3调控一系列与癌细胞生存、增殖、血管生成、侵袭、转移、耐药性和免疫逃避有关的基因。在许多恶性肿瘤中检测到组成型活性STAT3,包括乳腺癌、黑色素瘤、前列腺癌、头颈鳞状细胞癌 (HNSCC)、多发性骨髓瘤、胰腺癌、卵巢癌和脑瘤。因此,靶向STAT3信号通路已成为许多癌症的有希望的治疗策略。
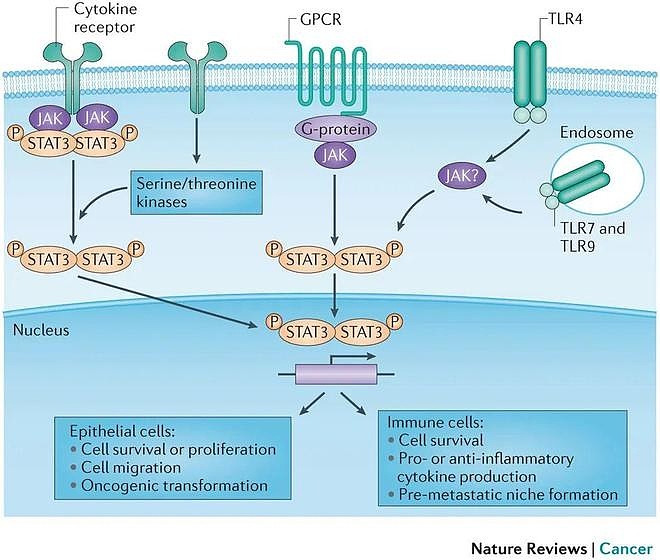
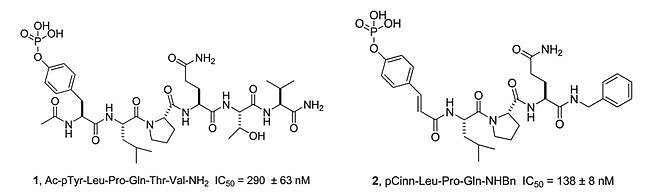
JAK-STAT3信号转导通路在肿瘤中的作用 STAT3也与一些自身免疫疾病有关,例如类风湿性关节炎、系统性红斑狼疮、多发性硬化和干燥综合征等。这些自身免疫疾病的特征是机体对自身组织或器官产生免疫反应,导致组织损伤和功能障碍。STAT3在这些自身免疫疾病中的作用主要是通过影响Th17细胞的分化和功能,以及调节T细胞和B细胞的平衡。Th17细胞是一种促炎性的T辅助细胞亚群,它可以分泌白介素17 (IL-17)等促炎因子,参与多种自身免疫性炎症反应。STAT3是Th17细胞分化的关键转录因子,它可以被IL-6等细胞因子激活,从而诱导RORγt等基因的表达,促进Th17细胞的产生。 另一方面,STAT3也可以抑制T调节细胞 (Treg)的分化,Treg是一种具有免疫抑制功能的T细胞亚群,它可以通过分泌白介素10 (IL-10)等抗炎因子,抑制自身免疫反应。此外,STAT3还可以影响B细胞的活化和抗体的产生,进一步加剧自身免疫性损伤。因此,STAT3在自身免疫疾病中起着重要的促进作用,靶向STAT3的抑制剂或降解剂可能是治疗这些疾病的有效策略。 目前已经发现了许多抑制或降解STAT3活性的化合物,它们可以分为两大类: 抑制STAT3上游信号或直接作用于STAT3蛋白本身。抑制上游信号的方法主要是通过抑制JAK、Src或受体酪氨酸激酶(RTK)等分子来阻断STAT3的磷酸化。直接作用于STAT3蛋白的方法主要是通过干扰SH2、DBD、NTD或Tyr705等结构域来阻断STAT3的二聚化、核转位或DNA结合 。其中一些化合物已经进入临床试验阶段,如JAK抑制剂ruxolitinib、Src抑制剂saracatinib和小分子SH2抑制剂C188-9等。这些化合物在一些癌症模型中显示出抗肿瘤效果,但也存在一些局限性,如水溶性差、细胞通透性低、选择性不高、毒副作用大等。因此,开发更有效、更安全、更特异的STAT3靶点化合物仍然是一个重要的研究方向。 STAT6主要由细胞因子白细胞介素-4和白细胞介素-13激活。STAT6在肿瘤和自身免疫中有重要的作用。STAT6可以调节B细胞分化、抗体类别转换、T细胞极化、巨噬细胞激活、肥大细胞增殖和分泌等免疫反应。STAT6的异常表达或激活与多种肿瘤的发生和发展有关,如孤立性纤维瘤、乳腺癌、前列腺癌、黑色素瘤、恶性淋巴瘤等。STAT6也参与了一些自身免疫性疾病的发病机制,如哮喘、类风湿性关节炎、系统性红斑狼疮等。 STAT6是一种有价值的肿瘤诊断标志物和治疗靶点,也是自身免疫性疾病的潜在干预对象。STAT6的检测可以用于区分孤立性纤维瘤(阳性染色)和其组织学模拟物(阴性染色)。STAT6抑制剂可以用于治疗依赖于STAT6信号通路的肿瘤和自身免疫性疾病。 磷酸肽类Warhead的由来 STAT3通过其SH2结构域被募集到磷酸化受体。然后,它在Tyr705上被JAK激酶、Src、Abl或受体的激酶活性磷酸化。酪氨酸磷酸化后,Stat3形成二聚体,其中一个蛋白质分子的SH2结构域与另一个的pTyr705残基结合,反之亦然。二聚体迁移到细胞核,与特定的DNA序列结合并启动转录。相比于间接抑制上游亦或是抑制DBD,NTD,直接靶向SH2抑制STAT3二聚化显然是最优的选择。 鉴于STAT3通过其SH2结构域与gp130、白血病抑制因子受体(LIFR)、表皮生长因子受体(EGFR)、白细胞介素10受体(IL-10R)、和粒细胞集落刺激因子受体(G-CSFR)上的磷酸酪氨酸残基结合。对STAT3抑制剂的早期探索便尝试通过合成和测试来自不同受体结合位点的磷酸化肽,寻找抑制Stat3受体结合和二聚化的先导肽。研究发现Y(p)LPQTV是最优的磷酸化肽序列,它来自gp130的Tyr904位点,具有290 nM的IC 50 值,比来自STAT3自身的磷酸化肽序列高133倍。
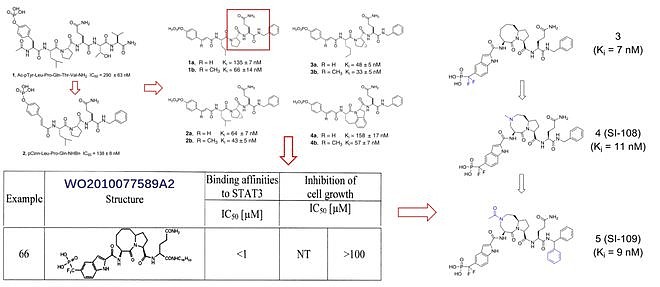
第一代与第二代GPR130 Tyr904位衍生磷酸肽 后续不作细说,经过德克萨斯大学John S. McMurray团队与密歇根大学王少萌团队多年的改造优化(主要是烯基部分与苯环扣五元杂环,以及吡咯环处扣八元环),这类磷酸肽类抑制剂与STAT3的亲和力达到了低纳摩尔水平。
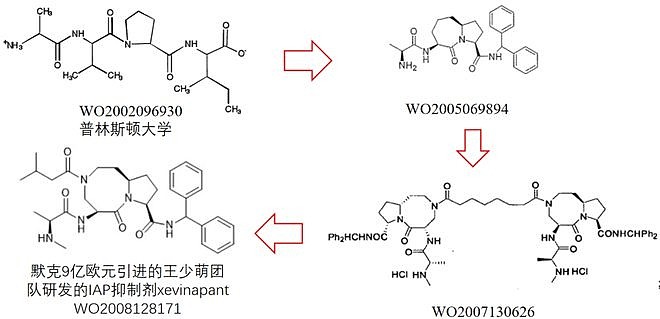
在化合物改造上王少萌团队可谓功不可没,这一关键性母核五元并八元环结构的出现可以追溯到其更早的XIAP专利WO2005069894与WO2007130626。(最早的原型WO2002096930的XIAP抑制剂结构与这类磷酸肽类抑制剂很相似,也许从这里得到的灵感,将此母核结构挪用到了STAT3抑制剂上)。
然而这类磷酸肽类抑制剂也普遍存在一些问题。如王少萌2010年在ACS Med. Chem. Lett.发表的文章所描述的那样,尽管这类化合物具有出色的STAT3亲和活性,但却没有细胞活性。
出现这种情况可能是由于这类分子糟糕的成药性(头部磷酸基团与末端谷氨酰胺导致),导致其难以透过细胞膜。似乎对磷酸肽类STAT3抑制剂的研究走到了一条死胡同,对此的研究也停滞不前,直到近几年有了新的突破,对此的研究走向了两条不同的道路。 磷酸肽类STAT3 PROTAC PROTAC技术想必不用多说,其最大的特点也是最吸引人的一点便在于,其warhead仅仅只需要与靶蛋白有足够的亲和力,不需要抑制活性便能够降解靶蛋白。从而使其能够降解一些以往认为是“undruggable”的靶点,达到疾病治疗的目的。这一策略似乎是对STAT3量身定制的,毕竟这类磷酸肽类抑制剂只有亲和力而缺乏抑制活性。而STAT3目前也是PROTAC领域进入临床阶段的针对“undruggable”靶点最好的佐证。 根据NextPharma数据库相关信息显示,目前以磷酸肽类抑制剂作为Warhead的STAT3 PROTAC研究机构主要有密歇根大学(王少萌),Kymera,以及国内的和正医药(CYD0618、PW0965与PW1087的warhead是冬凌草甲素衍生物)。
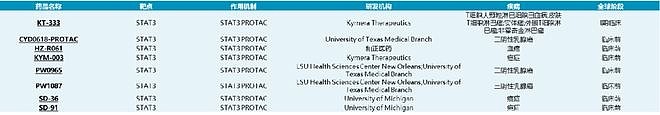
通过NextPat数据库,对这三家公司前沿专利检索整理后如下表:
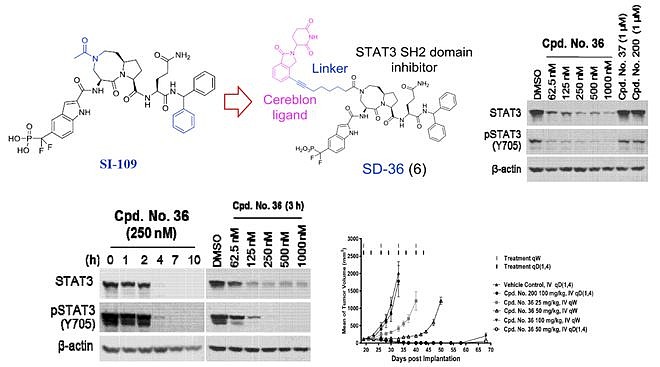
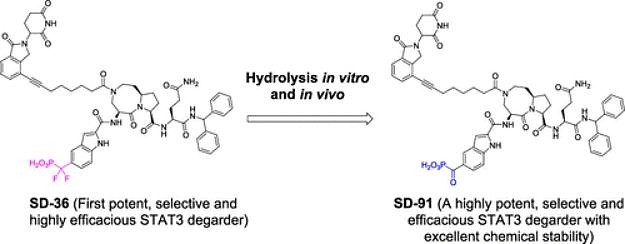
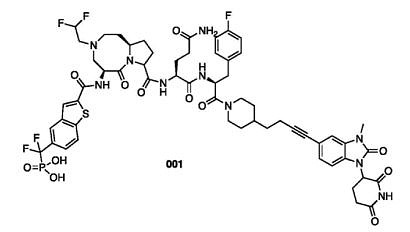
首先介绍一下王少萌的STAT3 PROTAC专利,共三篇。 WO2020198435,SD-36的相关专利。Warhead是文章中提到的化合物SI-109,E3连接酶选择CRBN,Linker多为炔基脂肪链,linker链接位点为母核大环N上或是磷酸肽类末端苯环上。活性均已在文章中有所体现。列举结构与部分活性数据。
结合文章(ACS Med. Chem. Lett. 2021, 12, 6, 996–1004)与专利,还提到化合物SD-91(专利中Cpd.92),是SD-36的水解产物,它能够高亲和力地结合到STAT3蛋白上,并且对其他STAT家族蛋白成员显示出超过300倍的选择性。
WO2020205467,与第一篇专利几乎同时申请。区别在于该专利对SI-109进行了进一步的改造,如改变母核大环结构,或探索新的Linker连接位点。但由于该专利缺乏活性数据,因此不作过多评价。列举个别代表性化合物。
WO2021195481为第三篇专利,本专利在Linker的选择上,部分保留了上两篇中采用的炔基脂肪链结构,也尝试引入目前较为成熟的、成药性更好的脂肪杂环链。同时,还尝试了将磷酸基团酯化做成前药。由于专利中化合物对STAT3降解并不出色,且对STAT1缺乏选择性(抑制STAT1会有抑制免疫,肿瘤恶化,内分泌紊乱等潜在副作用),本专利价值不高。
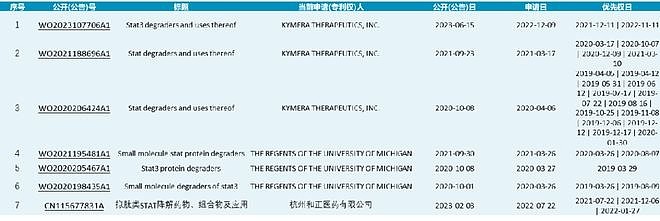
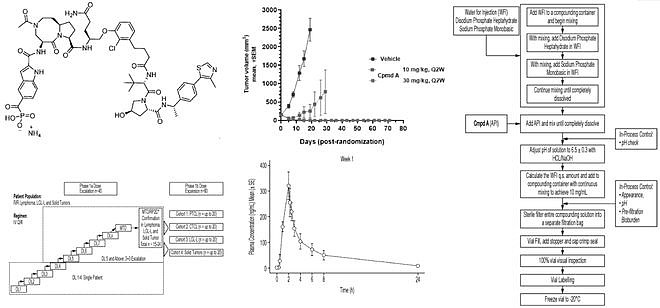
下面讲一下Kymera的STAT3 PROTAC专利,共三篇。 Kymera在STAT3 PROTAC上做了广泛的研究,涉及疾病领域包括血液瘤,实体瘤,以及自身免疫疾病(临床前数据详见公司官网)。其管线KT-333项目也于2022年进入I期临床研究,并于今年公布了初步的临床数据。结果表明:KT-333在外周血单核细胞中实现了高达88%的平均最大STAT3降解,有STAT3通路抑制(SOCS3减少)和外周血中炎症生物标志物下调的证据;最常见的治疗引起的不良事件为1-2级疲劳、贫血和胃肠道症状,没有DLT或药物相关的SAE;Ia期剂量递增正在进行中,并继续入组至剂量水平4(0.4 mpk)。 Kymera对STAT3 PROTAC化合物改造主要是专利WO2020206424与WO2021188696。在这两篇专利中,Kymera尝试了不同形式的Warhead(部分同SD36,部分同SD-91,也做了磷酸酯前药),不同的Linker类型与连接位点,同时在E3连接酶选择上选取了CRBN与VHL两种。 对结构改造部分不作细说,因为在其最新公开的专利WO2023107706中,公开保护了化合物A(即WO2020206424中的Cpd.233)的盐型、液体制剂(API)、适应症,临床给药方案,以及I期的初步药代数据。这些证据应该能够表明,这个CompoundA,极有可能便是临床化合物KT-333。
和正医药的STAT3 PROTAC项目HZ-R078于今年AACR上以Poster的形式公开,数据显示其具有不错的体内外抗肿瘤活性与药代参数。目前正在往IND的方向推进。其专利CN115677831可讲的不多,从专利时间轴上看,最早优先权是2021.07.22,在王少萌与Kymera专利公开之后,想必化合物设计上对两者均有借鉴。事实上也确实如此,从该专利实施例中看,其化合物设计思路便是对两者专利的拼接。代表化合物如下:
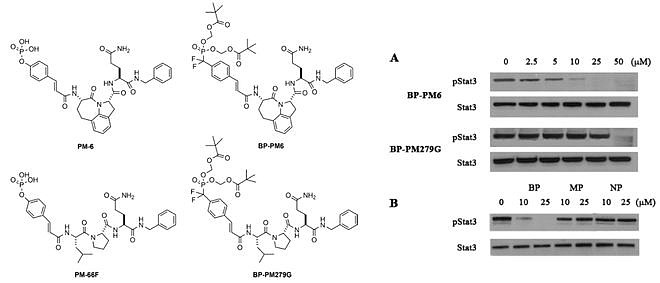
经过二十多年的发展,PROTAC技术愈发成熟,目前在研的PROTAC药物披露的临床数据释放出积极的安全性和疗效信号,同时愈来愈多的PROTAC药物进入临床开发阶段。上述这类基于磷酸肽类抑制剂的STAT3 PROTAC算是一个“柳暗花明”的发现,尽管由于潜在成药性问题未进入临床阶段,SD-36也成为了首个靶向STAT3的PROTAC分子,对PROTAC技术降解“undruggable”靶点提供了良好的佐证。KT-333虽然由于成药性问题只能作为液体制剂,但是作为首个进入临床阶段的STAT3 PROTAC,其临床结果已初有成效。相信其今后会有一个好的结果。 口服磷酸肽类STAT抑制剂 2009年似乎是磷酸肽类抑制剂研发的分水岭。王少萌认为这条路似乎走不通,选择了蛰伏近十年直到PROTAC技术的出现。而John S. McMurray选择了走向另一条道路:做磷酸酯前药。 该团队发现阻碍基于磷酸肽的SH2结构域抑制剂开发的两个主要挑战是磷酸基团的负电荷,它阻止了细胞膜上的被动扩散,以及磷酸基团对磷酸酶活性的不稳定,这使得磷酸肽无法识别。此外,酪氨酸的磷酸酯很容易被磷酸酶裂解,从而破坏抑制剂与SH2结构域结合的能力。磷酸盐、二氟甲基膦酸酯、丙二酸酯和杂环化合物已被用作磷酸酯替代物来克服这一缺陷。作者认为这些前药在进入细胞后可以被羧酯酶水解,释放出芳香二氟甲基磷酸盐,与STAT3的SH2结构域结合,从而分解已形成的STAT3二聚体,并阻断STAT3与受体的结合,从而阻止其后续基因的表达。 进一步的研究发现,双磷酸酯前药BP-PM6具有细胞活性,能抑制STAT3的磷酸化,尽管活性较差。而单磷酸酯前药(MP)与磷酸(NP)并没有活性。(基于WO2010118309与Organic letters, 2009, 11(15): 3394-3397)
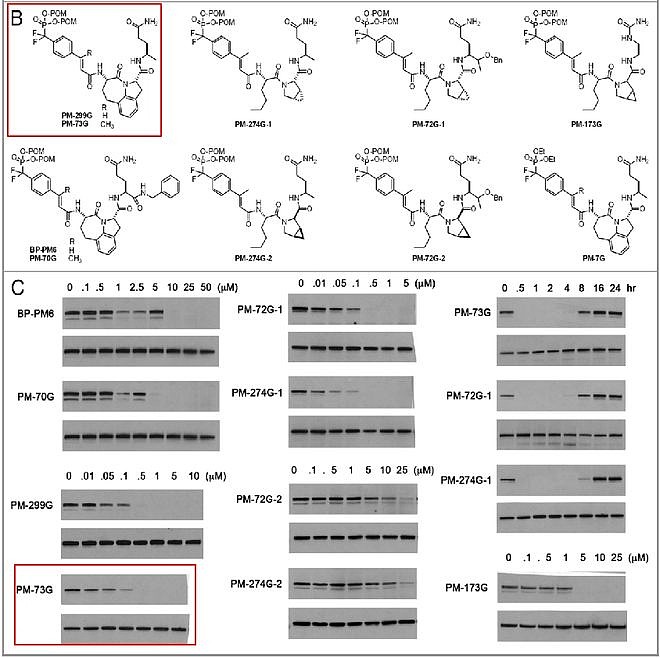
这一鼓舞人心的结果使得该团队对此进行进一步的优化,化合物PM-73G可以在低纳摩尔浓度下显著抑制STAT3的磷酸化。
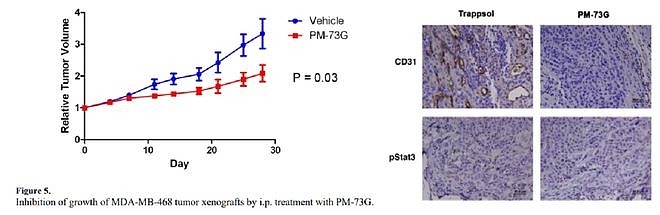
紧接着该团队便尝试探究PM-73G的体内抗肿瘤活性,令人失望的是,尽管该化合物在体内很好的抑制了肿瘤内STAT3的磷酸化,但并没有一个很好的肿瘤生长抑制效果(近年来也有种种证据表明,STAT3 727位点磷酸化引导的非经典信号通路在肿瘤发生发展中起着举足轻重的作用,仅抑制STAT3的Tyr705并不足以达到抑制肿瘤的效果。这是后话,这里不作细说)。
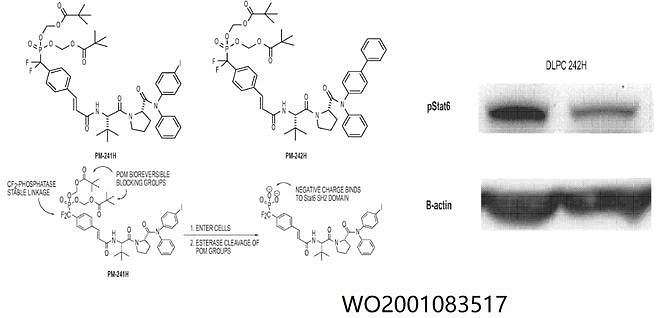
利用磷酸肽类STAT3抑制剂治疗肿瘤似乎走到了尽头,John S. McMurray团队在寻求新的母核的STAT3抑制剂的同时,把研究方向指向了另一处:STAT6。 Tularik公司(已被Amgen收购)于1999年构建了一类STAT4/STAT6抑制剂的高通量筛选方法(WO1999050283),并于2001年公开了STAT4/STAT6二肽衍生物专利(WO2001083517)。这类磷酸肽类前药能够在进入细胞水解成磷酸盐,抑制STAT6的磷酸化,但活性较差,在几十微摩尔水平。与STAT3抑制剂不同的是,这类结构除了通过前药解决磷酸基团弊端外,末端也没有类似的谷氨酰胺结构,这一点大大提升了这类分子的成药性。
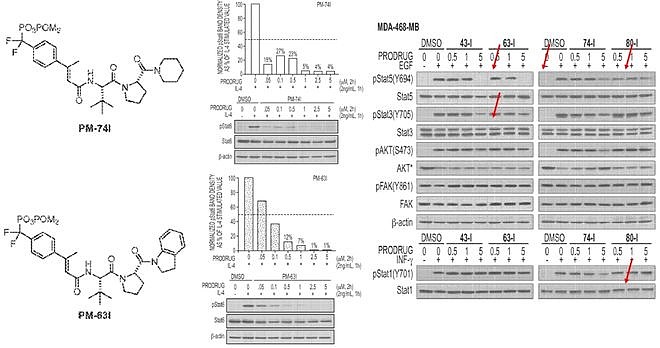
John S. McMurray团队以此为基础,在PM-242H基础上作了进一步的结构改造与优化。这份工作(WO2014182928)也取得了不错的成果,多数化合物对STAT6的磷酸化抑制水平达到了几十纳摩尔的水平。
在这之后,John S. McMurray团队发了几篇文章,阐述了专利中这类药物的相关生物学机制。其中2015年发表于J Med Chem的文章表明:STAT6是受IL-4和IL-13调节的转录因子,在过敏性气道疾病中起重要作用。作者开发了一种由磷酸肽模拟物组成的化合物,这些模拟物可以通过STAT6的SH2域与其结合,并阻止其与IL-4或IL-13受体上的磷酸化酪氨酸残基相互作用。这导致STAT6的Tyr641磷酸化和转录活性被显著抑制。这些结果表明,靶向STAT6 的SH2域是一种有希望治疗过敏性气道疾病的方法。 在2018年的针对炎性乳腺癌的文章表明:IL-4和IL-13通过STAT6 Tyr641磷酸化来促进巨噬细胞的M2极化。STAT6 Tyr641磷酸化是M2极化所必需的。磷酸肽类抑制剂PM37能够特异性地抑制STAT6的SH2结构域,阻断STAT6 Tyr641磷酸化,从而阻断IL-4/IL-13/IL-4受体α(IL-4Rα)信号通路,降低M2型TAM的极化水平。并通过小鼠模型证明了PM37可以有效地增强炎症性乳腺癌细胞对放射治疗的敏感性,并且可以逆转已经存在的放射抵抗性。同时PM37可以通过下调蛋白激酶Cζ(PRKCZ)来抑制M2型TAM诱导的IBC细胞放射抵抗性。因此,本研究揭示了IL-4/IL-13/STAT6信号通路在调节巨噬细胞极化和影响IBC放射反应中的重要作用,并且提出了一种通过干扰该信号通路来增强IBC放射敏感性的新策略。 2018年的另一篇文章介绍了STAT5/6双靶点磷酸肽类抑制剂PM-43I,通过对PM-43I进行初始剂量范围、毒性和药代动力学实验,发现它能有效抑制小鼠中依赖STAT5和STAT6的过敏性气道疾病。此外,PM-43I以最低有效剂量0.25μg/kg逆转了小鼠预先存在的过敏性气道疾病。值得注意的是,PM-43I通过肾脏高效清除,没有长期毒性。PM-43I代表了一类可能适合进一步临床开发用于治疗哮喘的小分子药物。 然而在这之后John S. McMurray团队并未在此基础上更进一步,在此之后并未有新的相关专利公开。不过专利中的数据体现出了另一个有趣的结果:这类STAT6磷酸肽类抑制剂,在保持其他部分不变的情况下,只改动末端基团,能够做出对STAT家族的不同选择性(PM-43I对STAT5与STAT3均有一定的活性)。 无独有偶,在今年王少萌发表于Nature Chemical Biology的文章中,对其发现的一类磷酸肽类抑制剂做了结构改造(部分化合物来自John S. McMurray的上述专利WO2014182928,想必也是从中找到的灵感),将一类选择性STAT6抑制剂改造成STAT5/6双靶点抑制剂,再以此为warhead,设计成STAT5 PROTAC。
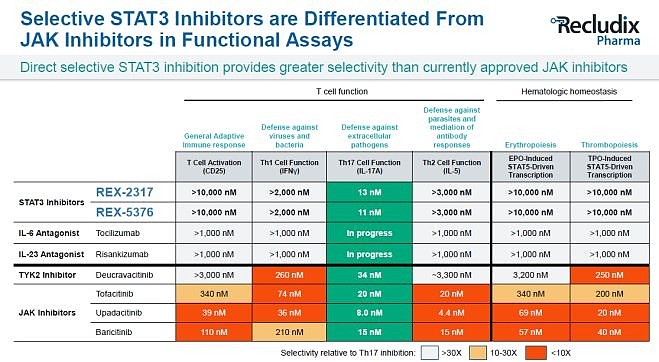
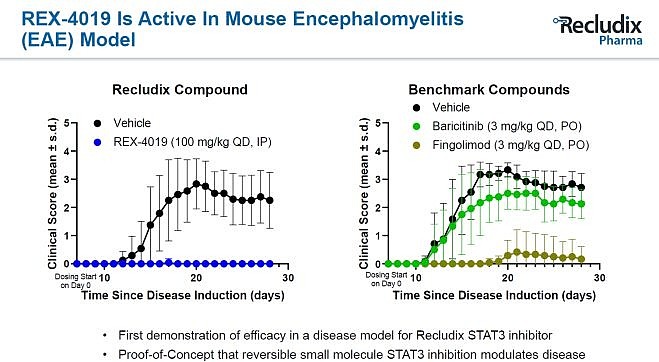
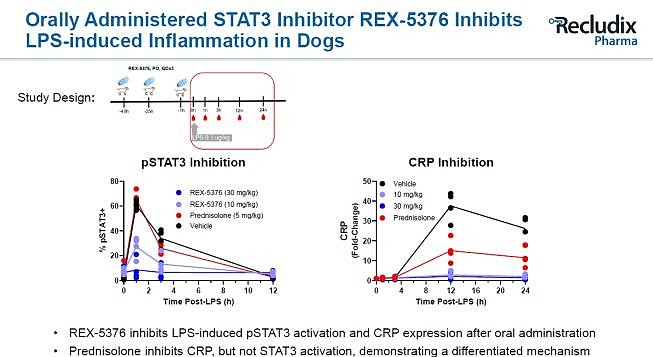
这些结果带来了一种假设:是否能够在此基础上进一步优化,最终设计出针对STAT家族单一靶点的高选择性且高亲和力的磷酸肽类抑制剂? Recludix Pharma给我们揭晓了答案。这家公司于2021年完成6000万美元A轮融资,旨在针对SH2结构域开发精准小分子药物,最初的重点是STAT3、STAT6。从Cooperation Presentation与公开专利中可以看到,Recludix想要开发一类口服的磷酸肽类STAT3/6抑制剂,并选择了自身免疫疾病作为适应症开发。 针对STAT3项目,他们公开了化合物REX-2317,对STAT3磷酸化抑制活性达到了5.2nM。且REX-2317对Th17炎症细胞抑制活性比肩JAK/TYK2抑制剂,选择性媲美IL-6/IL-23单抗。另一个STAT3抑制剂REX-5376也以口服给药的形式,在LPS诱导的狗炎症动物模型中表现出优异的炎症抑制效果。不过这类化合物还存在一些问题,REX-2317对STAT1的选择性仅仅只有12倍,这可能会导致一些潜在的毒副作用。
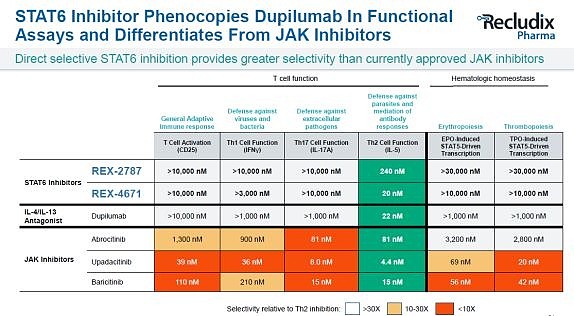
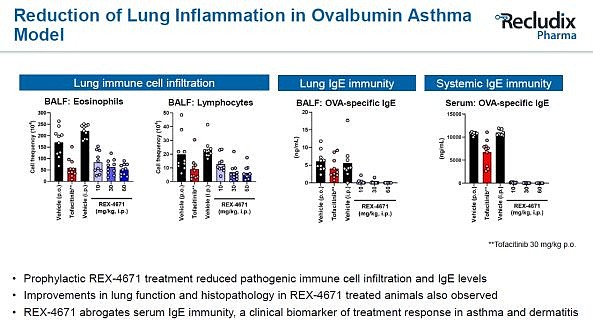
Recludix对STAT6项目似乎更有信心。在更早些的JPM会议Presentation中,Recludix声称其STAT6抑制剂有望成为“口服达必妥”(即赛诺菲针对IL-4的度普利尤单抗,去年销售额约83亿美元,预测销售峰值将达到140亿美元)。目前公开的STAT6抑制剂REX-2787的STAT6磷酸化抑制活性达到了16nM,且具有较高的选择性。与STAT3抑制剂类似,这类STAT6抑制剂对Th2炎症细胞抑制活性比肩JAK/TYK2抑制剂,选择性媲美IL-4/IL-13单抗。此外,这类STAT6抑制剂的体内药效也在哮喘动物模型中得到了验证,且优于已上市JAK抑制剂托法替尼(不过给药方式是腹腔注射,可能还在优化,未达到能够口服的成药性)。
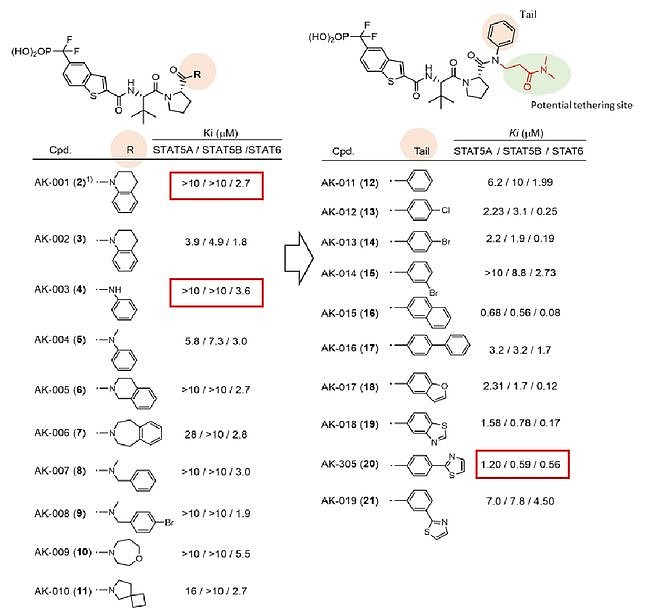
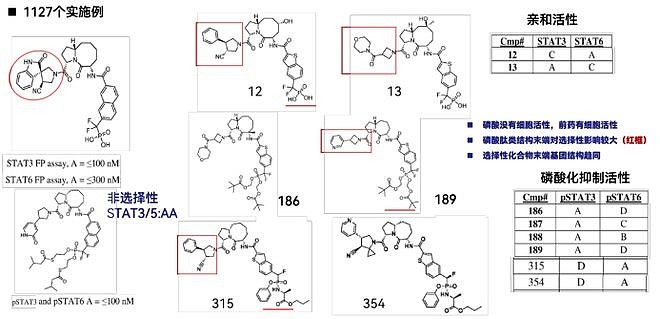
整体进度上看,Recludix预期分别在今明两年拿到STAT3与STAT6的PCC,并分别在2024年与2025年将STAT3与STAT6抑制剂推向I期临床。 那么,这类STAT小分子抑制剂到底长什么样呢?从最新公开的专利中,我们可以看到他们的工作成果,我对此的评价是:博采众长,踵事增华。这类化合物具有与王少萌专利WO2010077589相同的磷酸肽母核结构,并在John S. McMurray的专利WO2014182928基础上,对末端基团的多样性做出了进一步的探索。最终找到了一类高选择性且具有高活性的STAT3与STAT6磷酸肽类小分子抑制剂。 WO2023133336,Recluix公开的第一篇专利,共1127个实施例。专利中公开了一系列不同选择性的STAT3/STAT磷酸肽类抑制剂与其前药形式。专利中的数据进一步证明了这类化合物只有前药具有细胞活性,且末端基团对STAT家族的选择性影响较大。同时,具有选择性的化合物结构末端基团具有相似的结构。且该基团的微小变化便能引起活性与选择性的巨大差异。
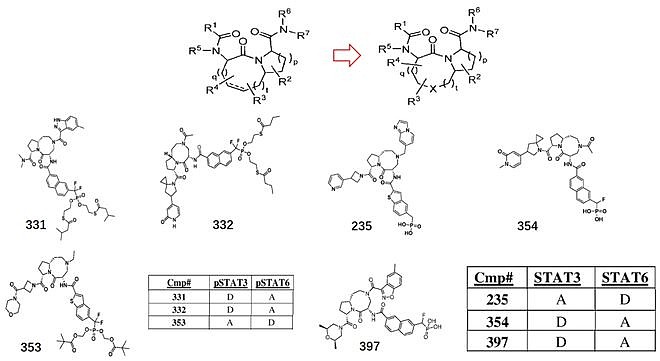
说句题外话,在这篇专利公开后没多久,传出来赛诺菲以1.25亿美元的首付款,12亿美元的里程碑付款的形式,与Recludix达成战略合作的消息,两家公司将共同开发和商业化全球首创的自免新药STAT6抑制剂,用于免疫和炎症疾病患者。据悉该合作是源于今年1月JPM大会上Recludix的汇报使赛诺菲产生了兴趣,通过长时间的谈判促成了此次合作。赛诺菲此举显然是想巩固其在Th2炎症疾病领域的优势地位。 WO2023164680,Recluix公开的第二篇专利,共441个实施例。从实施例中可以发现,Recludix在第一篇基础上在中心母核上引入N原子(与SI-109相同),并在此基础上探究取代基对活性的影响。同时对末端取代基作出进一步的探索,最终发现了一系列新的选择性STAT3与STAT6磷酸肽类小分子抑制剂。
后记 在2022年,cell上发表了一篇重磅综述,介绍了自1992年JAK-STAT信号通路被发现后的三十年来,JAK-STAT这一重要通路领域的研究概况。过去三十年针对这一领域有超过九万篇论文发表。针对JAK这个靶点,美国FDA已批准十几个JAK抑制剂。仅在2022年,JAK抑制剂的销售额达到了约100亿美元。但在JAK抑制剂大放异彩的当下,由于表面平坦缺乏结合口袋,STAT这个一直以来被认为的难成药靶点却始终都是药学界难以逾越的天堑。
过去三十年中,全球无数课题组与医药公司对STAT这座高峰发起冲锋,却纷纷铩羽而归。自1999年磷酸肽类抑制剂被发现以来,Tularik,John S. McMurray,与王少萌渐渐奠定了成功的基石。随后王少萌与Kymera把矛头指向了STAT3 PROTAC;而John S. McMurray与Recludix剑指磷酸肽前药抑制剂。两条不同的道路,最终在Kymera与Recludix上看到了希望的曙光。 与STAT齐名的另一个难成药靶点是KRAS,在过去也一直被认为不可成药,但随着2015年Araxes的ARS-1620的出现、后续KRAS G12C抑制剂开发全球的高度内卷、AMG510与MRTX849的先后上市,以及后来KRAS G12D抑制剂、KRAS G12V抑制剂,乃至Pan-KRAS与Pan-RAS抑制剂的先后发现,KRAS正慢慢摘掉其“undruggable”的帽子。随着Kymera的STAT3 PROTAC与Recludix的口服磷酸肽抑制剂的出现,相信之后会有更多类似于和正医药的跟随者,向着STAT发起冲锋,使得STAT与KRAS一样,揭下其“undruggable”的标签。 最后说一句,随着互联网的发展,医药领域也进入了信息大爆炸的时代。专利、文献、学术会议、以及投融资等信息,构成了一张完整的医药情报网。同时,专利文献包含了世界科技技术信息的90%~95%,也是医药情报网上最基础乃至最关键的一个环节。本篇文章便是顺着专利脉络,结合文献,投融资信息,对磷酸肽类STAT抑制剂的发展做出的梳理,希望读者能有所启发。当然本人个人能力有限,文中可能会有所遗漏,还请大家批评指正。 参考文献: 1.Bioorg. Med. Chem. Lett. 13 (2003) 633–636 2.J. Med. Chem. 2009, 52, 6126–6141 3.Organic letters, 2009, 11(15): 3394-3397. 4.ACS medicinal chemistry letters, 2010, 1(2): 85-89. 5.J. Med. Chem. 2011, 54, 3549–3563 6.Jak-Stat, 2012, 1(4): 263-347. 7.J Exp Ther Oncol. 2012;10(2):155-62. 8.International journal of peptide research and therapeutics, 2013, 19: 3-12. 9.Cancer cell 36.5 (2019): 498-511. 10.J. Med. Chem. 2015, 58, 22, 8970–8984 11.International Journal of Radiation Oncology* Biology* Physics 100.4 (2018): 1034-1043. 12.Journal of Biological Chemistry, 2018, 293(26): 10026-10040. 13.Nature Chemical Biology, 2023: 1-9. 14.J. Med. Chem. 2023, 66, 2717−2743 $海创药业-U(SH688302)$ $海思科(SZ002653)$ $亚盛医药-B(06855)$ 国内生物医药产业链相关上市公司 CXO:药明康德、凯莱英、泰格医药、昭衍新药、康龙化成、药明生物等; 生物制品:智飞生物、万泰生物、长春高新、沃森生物、华兰生物、甘李药业等; 化学制药:恒瑞医药、复星医药、华东医药、新和成、人福医药、科伦药业、信立泰等; 中药:片仔癀、云南白药、同仁堂、白云山、以岭药业、太极集团、济川药业等; 医院及诊断服务:爱尔眼科、通策医疗、金域医学等; 科学服务:诺唯赞、百普赛斯、优宁维; 医美:爱美客、华东医药、ST美谷、华熙生物等; 医疗设备:迈瑞医疗、联影医疗、微创医疗、乐普医疗、鱼跃医疗、九安医疗等; 医药商业:上海医药、益丰药房、大参林、九州通、国药一致、中国医药、海王生物等; 部分美股医药上市公司 礼来、诺和诺德、强生、默沙东、艾伯维公司、阿斯利康、诺华制药、辉瑞等 @价值at风险 @张翼轸 @丹书铁券 @今日话题@润哥 @微进化ing |

【本文地址】