| 计算能带自动产生k | 您所在的位置:网站首页 › vaspkit处理dos › 计算能带自动产生k |
计算能带自动产生k
|
今天主要记录强关联电子体系+U、DOS和能带计算过程中参数的设置,相信大家在读一些计算类文献时会看到文章中计算部分有The GGA + U calculations were performed using the model proposed by Dudarev et al 这样一段话,那么我们今天来讨论一下。 首先了解一下什么是强关联电子体系,对于弱关联体系,DFT-LDA近似就能很好的描述材料的电子特性。而对于含有d层尤其是f层电子的体系,电子--电子库伦互作用导致的局域电子占据态会强烈影响体系的能级分布。 电子间的库伦交互作用不可忽略---强关联体系。强关联体系的处理---引入Hubbard U模型: 就是LDA的总能,后面一项为考虑了电子库伦交换作用的Hubbard U项。 1) VASP计算过程LDA+U模型如何设置参数?U值由三个参数控制:LDAUL LDAUU LDAUJ LDAUL---对具体的p-/d-/f-轨道加U LDAUU---电子库伦相互作用项(on -site Coulomb interction) LDAUJ---电子交换相互作用项(on -site exchange interction) (U-J为有效U值) 2) U值怎么确定? 对U值做一系列测试,求得与实验值较为接近的结果。(纯计算)研究较为成熟的体系,参考文献值,然后引用出来即可。(主实验,辅计算)谁让我爱你们呢,详见:http://blog.sciencenet.cn/blog-567091-775079.html 光说不练瞎嚯嚯,上才艺: 一、CeO2晶胞结构优化首先找到CeO2结构 
POSCAR: CIF file1.0 5.4109997749 0.0000000000 0.0000000000 0.0000000000 5.4109997749 0.0000000000 0.0000000000 0.0000000000 5.4109997749 O Ce 8 4Direct 0.250000000 0.250000000 0.250000000 0.750000000 0.750000000 0.750000000 0.750000000 0.750000000 0.250000000 0.250000000 0.250000000 0.750000000 0.750000000 0.250000000 0.750000000 0.250000000 0.750000000 0.250000000 0.250000000 0.750000000 0.750000000 0.750000000 0.250000000 0.250000000 0.000000000 0.000000000 0.000000000 0.000000000 0.500000000 0.500000000 0.500000000 0.000000000 0.500000000 0.500000000 0.500000000 0.000000000INCAR: #### initial I/O ####SYSTEM = CeO2ISTART = 0 ICHARG = 2 LWAVE = .F. LCHARG = .F. #### Ele Relax #### ENCUT = 400 ISMEAR = 0 SIGMA = 0.2 EDIFF = 0.1E-4 NELM = 100VOSKOWN = 1 #PW91泛函设置为1,PBE或PBEsol函数不需要设置 LREAL = .F.PREC = NormalALGO = Fast#### Geo opt ####EDIFFG = -0.05IBRION = 2 POTIM = 0.5 NSW = 100 ISIF = 3 #### Mag ####ISPIN = 2 #CeO2有spin电子### +U ###LDAU = .TRUE. #(打开+U设置,控制计算中是否考虑在位库伦较正相)LDAUTYPE = 2 #(+U的类型,1/2/4;2为默认值,U值由LDAUU减去LDAUJ确定)LDAUL = -1 3 # (控制具体轨道上加U;-1:不加U;1-p轨道;2-d轨道;3-f轨道)LDAUU = 0 5.5 #(O不加U,Ce的f轨道上+U,U值取5)LDAUJ = 0 0.5 LMAXMIX = 6 #(对+U日系,设置线性混合参数,f轨道设为6,d轨道设为4)如果不理解下面介绍认真看这里LDAUL、LDAUU、LDAUJ的格式是要参考POSCAR中有多少类型原子,这里CeO2的PODCAR中只有Ce和O原子两种原子,按照POSCAR中顺序O不+U所以LDAUL第一个值为-1,Ce的f轨道电子要+U所以LDAUL第二个值为3,同理LDAUU和LDAUJ的格式一样,O不+U所以LDAUU和LDAUJ第一个值都为0,那么LDAUU-LDAUJ=0;Ce的U值我要设为5所以LDAUU和LDAUJ第二个值分别为5.5和0.5,那么LDAUU-LDAUJ=5。总体来说U值的设置按照你POSCAR中原子顺序来,第一种类型需要+U就设置LDAUL、LDAUU、LDAUJ的值,不需要就设置LDAUL=-1、LDAUU=0、LDAUJ=0,我再拿一个我自己算的体系看一下就透彻了: Ni V Rh H O 9 2 1 24 24 LDAUL= 2 2 2 -1 -1 LDAUU= 4 3.5 3.5 0 0 LDAUJ= 0 0 0 0 0 LMAXMIN= 4这个我应该表述的很明白了吧,KPIONTS中各个基矢方向上分割各基矢的点数4 4 4即可。 二、CeO2电子自洽计算Scf-INCAR: NSW = 0 #原子弛豫步数为0,离子固定不动IBRION = -1 #离子固定不动,有点多此一举,只要NSW=0了,IBRION其实不用变NELM = 200 #让电子充分自洽LWAVE = .T. LCHARG = .T. ISIF = 2其余参数保持不变, 再次提交POSCAR、POTCAR、INCAR、KPINTS和提交脚本计算即可。 计算完之后查看输出EIGENVAL文件,记住能带数(18) cat EIGENVAL 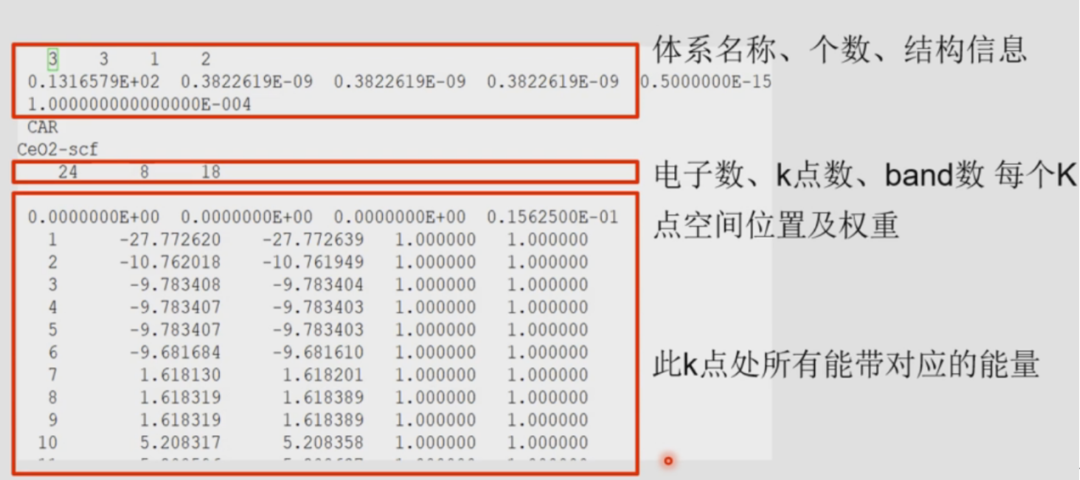 三、CeO2非自洽计算
三、CeO2非自洽计算
dos-INCAR: #### initial I/O ####SYSTEM = CeO2ISTART = 1 #(读入已有波函数文件)ICHARG = 11 #(读取前一步计算完的CHGCAR)LWAVE = .T. LCHARG = .T. #### Ele Relax #### ENCUT = 400 ISMEAR = -5 SIGMA = 0.2 EDIFF = 0.1E-4 NELM = 200VOSKOWN = 1 LREAL = .F.PREC = NormalALGO = Fast#### Geo opt ####EDIFFG = -0.05IBRION = -1 POTIM = 0.5 NSW = 0 ISIF = 2 #### Mag ####ISPIN = 2 #CeO2有spin电子### +U ###LDAU = .TRUE.LDAUTYPE = 2 LDAUL = -1 3 LDAUU = 0 5.5 LDAUJ = 0 0.5 LMAXMIX = 6### DOS ###NBANDS = 18 #默认值=NELECT/2+NIONS/2(没有自旋极化)=0.6*NELECT+NMAG(自旋极化)决定了在计算过程中带的真实数量。一般默认的就够,可不设置。LORBIT = 10 #将分波态密度输出到DOSCAREMIN = -35 #能量范围最小和最大值,从scf计算出的EIGENVAL文件中查找EMAX = 15NEDOS = 1000 #想让你的DOS图更顺滑,设置到2000也可以其余输入文件不变,把上一步电子自洽计算出的CHACAR和WAVECAR复制到一起提交计算即可。算完之后可用Vaspkit输出你想要的TDOS或PDOS,复制到Origin里作图即可,这里不做分析。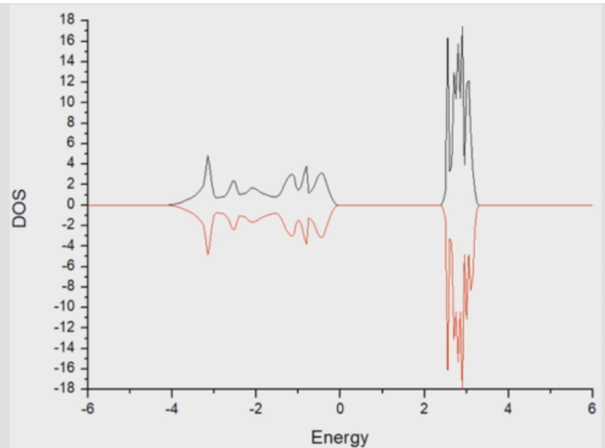 四、CeO2能带计算
四、CeO2能带计算
能带计算的话输入文件同上,需要修改的是KPOINTS文件,传统方法使用Materials studio需要人为的去确定K-path,并手动写入KPOINTS文件,非常麻烦而且易出错。使用seeK-path网站:则需要每次都把结构文件上传。而使用VASPKIT,只需要一个命令就可以在LINUX系统里完成这项工作,VASPKIT使用的生成K-path规则和seeK-path网站是完全一样的。而且VASPKIT的3-303/302/301功能不但能够处理3D材料,也可以处理2D(302功能)和1D材料。VASPKIT程序并不保证此功能的正确性,所以要对比此结果和seeK-path网站的结果( https://www.materialscloud.org/work/tools/seekpath) ①借助syml和gk.k两个脚本共同生成,根据scf计算的OUTCAR文件编辑syml文件(想要这些脚本的私聊):vi syml ②VASPKIT生成的KPOINTS文件(推荐): K-Path Generated by VASPKIT. #注释行 20 Line-ModeReciprocal 0.0000000000 0.0000000000 0.0000000000 GAMMA 0.5000000000 0.0000000000 0.5000000000 X 0.5000000000 0.0000000000 0.5000000000 X 0.6250000000 0.2500000000 0.6250000000 U 0.3750000000 0.3750000000 0.7500000000 K 0.0000000000 0.0000000000 0.0000000000 GAMMA 0.0000000000 0.0000000000 0.0000000000 GAMMA 0.5000000000 0.5000000000 0.5000000000 L 0.5000000000 0.5000000000 0.5000000000 L 0.5000000000 0.2500000000 0.7500000000 W 0.5000000000 0.2500000000 0.7500000000 W 0.5000000000 0.0000000000 0.5000000000 X③seeK-path网站生成的路径: Special k-points for band structure ! intersections line-modereciprocal 0.0000000000 0.0000000000 0.0000000000 1 GAMMA 0.5000000000 0.0000000000 0.5000000000 1 X 0.5000000000 0.0000000000 0.5000000000 1 X 0.6250000000 0.2500000000 0.6250000000 1 U 0.3750000000 0.3750000000 0.7500000000 1 K 0.0000000000 0.0000000000 0.0000000000 1 GAMMA 0.0000000000 0.0000000000 0.0000000000 1 GAMMA 0.5000000000 0.5000000000 0.5000000000 1 L 0.5000000000 0.5000000000 0.5000000000 1 L 0.5000000000 0.2500000000 0.7500000000 1 W 0.5000000000 0.2500000000 0.7500000000 1 W 0.5000000000 0.0000000000 0.5000000000 1 X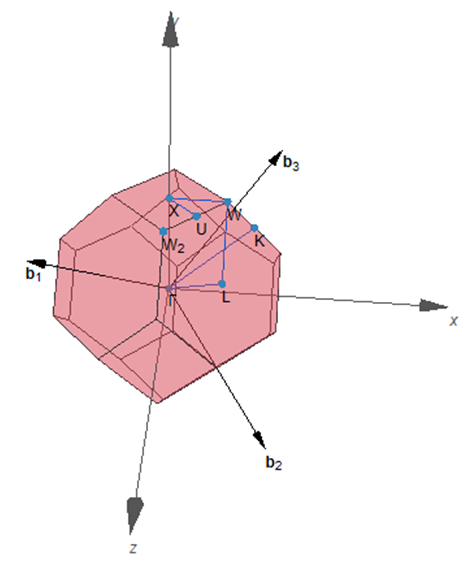
通过对比我们可以看到vaspkit和seeK-path网站生成的K-path路径是一样的。计算出的BAND.dat,BAND_REFORMATTED.dat保存了能带数据,可以直接导入Origin里画图,详见:http://blog.wangruixing.cn/2019/07/11/band2/ 今天就写到这吧,再写怕头发撑不住了。。。总结不易给个赞吧,你们的鼓励就是对我们最大的支持,爱你们么么哒! |
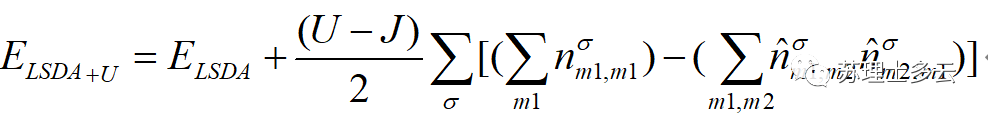
 Syml文件编辑完成后,运行gk.k文件:./gk.x则生成用于计算能带结构的KPOINTS文件:
Syml文件编辑完成后,运行gk.k文件:./gk.x则生成用于计算能带结构的KPOINTS文件:
【本文地址】