| Trycycler: consensus long | 您所在的位置:网站首页 › unicycle怎么读 › Trycycler: consensus long |
Trycycler: consensus long
|
方法和实现
Trycycler管道由多个单独运行的步骤组成(图中概述)。1,详见附加文件1:图S1)。在集群和协调步骤中,用户可能需要做出决策并进行干预。这意味着Trycycler不是适合于高吞吐量组装的自动化流程。Trycycler是用Python实现的,使用NumPy, SciPy和edlib包[23,24,25,26]. 图1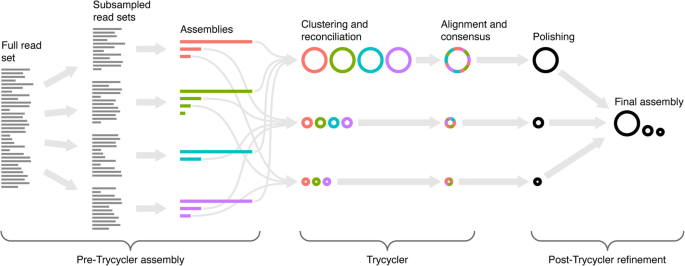
Trycycler长读组装管道概述。在Trycycler运行之前,用户必须生成相同基因组的多个完整集合,例如,通过组装原始长读集的不同子集。然后,Trycycler将来自不同程序集的contig聚类,并为每个簇生成一致的contig。然后可以对这些一致的contig进行抛光(例如,使用Medaka),并将其组合成最终的高质量长只读程序集 全尺寸图像在Trycycler运行之前,用户必须生成相同基因组的多个输入程序集(附加文件1:图S1A)。输入组件应该是完整的:每个复制子一个contig。如果无法实现完整的程序集(例如,由于读取长度不足)或读取深度较浅(例如,13].例如,在这里报告的测试中,我们使用Flye [10), Miniasm / Minipolish [13],乌鸦[11]和红豆[12].随机读子抽样可以提供进一步的独立性,其中每个程序集都是从完整读集的不同子样例生成的(Trycycler v0.5.0有一个“子样例”命令来实现这一点)。因此,更深层的长读集是可取的,因为它们支持更独立的子集。 Trycycler管道中的第一步是contig集群(附加文件1:图印地)。它的目的是将来自不同输入程序集的相同复制子的contig分组,因此后续步骤可以在每个复制子的基础上执行。例如,如果有问题的基因组有一个染色体和一个质粒,那么Trycycler聚类应该产生两个簇:一个用于染色体群,另一个用于质粒群。为了进行聚类,Trycycler对contigs之间的所有成对Mash距离进行完全链接层次聚类[27].为了帮助解释,FastME树使用了成对距离[28].在聚类完成后,用户必须判断哪些聚类是有效的(即,代表基因组中完全组装的复制子),哪些是无效的(即,代表不完整的、错误组装的或虚假的序列)——这是Trycycler过程中人类判断的关键。 下一步是“协调”每个集群的连续序列(附加文件1:图就是S1C)。这涉及到在必要时将序列转换为它们的反向补码,以确保集群中的所有序列都处于相同方向。大多数细菌复制子是圆形的,因此Trycycler将每个contig的开始和结束与集群中的其他contig对齐,以确定是否需要添加或删除碱基以实现干净的循环(对于线性复制子,可以使用—linear选项禁用)。然后它将每个序列从相同的位置开始旋转。某些基因序列(例如,dnaA和repA)通常作为全基因组的起始位置,因此Trycycler包含这些基因的数据库,并优先使用它们作为contig起始位置(参见“方法”一节)。如果没有从这个数据库中找到序列(≥95%的覆盖率和≥95%的一致性),Trycycler将使用一个随机选择的唯一序列代替。如果一个contig不能被循环,或者如果集群中的任何一对对齐具有较低的标识,则集群协调将失败。在这种情况下,Trycycler将建议采取干预措施来解决问题,但需要用户手动排除或修改contig序列。 调整后,每个簇的序列将有一个一致的链和起始位置,使它们适合全局多序列比对(附加文件1:图S1D)。为了提高计算性能,Trycycler对序列进行细分,使用1-kbp片段,必要时对每个片段进行扩展,以确保片段之间的边界不以重复区域开始/结束。它使用肌肉[29为每个片段生成一个多序列比对,然后将这些片段缝合在一起,生成一个完整的簇序列的单一多序列比对。然后,Trycycler将整个读集与每个contig序列进行比对,以便将其分配给特定的集群(附加文件1:图S1E)。 Trycycler管道中的最后一步是为每个集群生成一个共识序列(附加文件1:图S1F)。它通过将多个序列比对划分到有或没有任何变异的区域来实现这一点。对于所有存在变异的区域,Trycycler必须选择哪一种变异将进入共识。将最优变量定义为与其他变量总汉明距离最小的变量,这种方法更有利于更常见的变量。在两个变量之间出现平局的情况下,Trycycler将集群的读值与每种可能性进行对齐,并选择产生最大对齐得分的那个。,这是最符合阅读的变体。在多序列比对中,对每个变异区域取最优变异,得到聚类的最终Trycycler一致序列。 在Trycycler完成后,我们建议对其共识序列执行长读抛光(附加文件1:图S1G)。抛光不包含在Trycycler中,因为该步骤可以特定于所使用的长读测序技术,例如Medaka [30.ONT组件的抛光。如果有短阅读,短阅读润色(例如,与Pilon [31),以进一步提高装配精度。 Trycycler v0.3.3(用于在本手稿中生成程序集的版本)的代码和文档可在DOI 10.5281/zenodo.3966493上获得。Trycycler (v0.5.0)的当前版本可以在GitHub上找到(github.com/rrwick/Trycycler). 模拟读的性能在硅读模拟允许直接测试装配精度的基础事实:读取从一个参考基因组产生,读取被组装,并产生的组装被比较回原始参考序列。在这个分析中,我们模拟了来自RefSeq中10个最常见细菌物种的10个参考基因组的短读和长读2:引用)。我们用长-只读方法组装每个基因组(Miniasm/Minipolish [13],乌鸦[11], Flye [10]和Trycycler)、长读优先混合方法(Pilon [31),以及短读优先的混合方法(unicycle [21])。我们使用两个主要指标来量化每个装配的染色体序列的准确性:平均同一性和100-bp最差同一性(100-bp滑动窗口中观察到的最小同一性)。 只比较长时间读取的汇编程序(Flye, Miniasm/Minipolish和Raven),很明显Flye表现最好1:图S2)。这在Pilon用短读进行润饰之前都是正确的(平均身份Q41 vs Q38;平均最差100 bp-同一性95.8% vs 50.8-90.9%)和在Pilon抛光后(平均同一性Q57 vs Q42-Q55;平均最差100 bp同一性96.1% vs 50.8-95.7%)。因此,我们的主要结果排除了Miniasm/Minipolish和Raven,只留下性能最好的长读汇编程序:Flye。 数字2使用10个模拟的读集,显示每个方法的平均装配标识和100-bp的最差装配标识。在这两个指标中,Trycycler可靠地生产了比Flye更高质量的组件(平均身份Q51 vs Q41;平均最差100-bp同一性99.5% vs 95.8%)。这个结果也适用于长读优先的混合程序集,其中Trycycler+Pilon优于Flye+Pilon(平均标识Q74 vs Q57;平均最差100-bp同一性99.9% vs 96.1%)。unicycle的短读优先混合程序集的性能明显低于长读优先混合方法(平均标识Q25;平均最差100-bp同一性76.5%)。 图2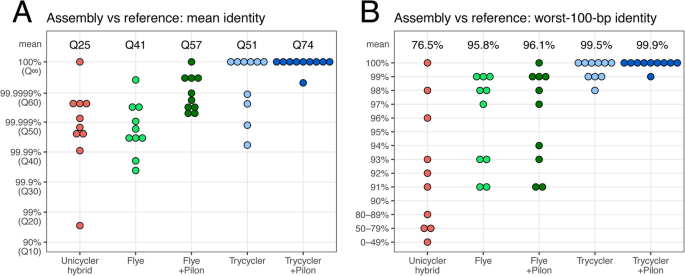
使用模拟读取的测试结果。对于10个参考基因组序列,我们分别模拟了短读和长读。然后使用Unicycler(短读优先混合程序集)、Flye(长读优先混合程序集)、Flye+Pilon(长读优先混合程序集)、Trycycler(长读优先混合程序集)和Trycycler+Pilon(长读优先混合程序集)组装读集。每条组装后的染色体与参考染色体重新对齐,以确定平均组装身份(一个)和100个bp滑动窗口中最差的身份(B).对于长只读组装,Trycycler始终比Flye实现更高的精度。Trycycler+Pilon(即使用Pilon以短读来抛光Trycycler组件)达到了最高的精度,比替代混合方法(unicycle和Flye+Pilon)做得更好。 全尺寸图像 实际读操作的性能由于模拟读取不能完全模拟真实排序[32,我们也用实读集测试了装配方法。我们为这项研究选择了7个细菌分离株(附加文件3.:基因组),每一种都属于不同的细菌物种,具有临床相关性。真正读取的挑战是缺乏一个明确的基础事实来比较集合。为了避免这个问题,我们为每个测试生物体生成了两组独立的长+短(ONT+Illumina)读数。简而言之,从每个生物体中提取一个DNA,用于制备两个ONT库(一个连接库和一个快速库)和一个Illumina库(其结果被分为两个不重叠的读集);详细资料载于“方法”一节。对于每种装配方法,我们比较了读集A和读集B的装配,它们之间的差异表明装配错误。虽然如果两个组合包含相同的错误,这种方法可能会出现假阴性,但它不会出现假阳性,因为只要同一基因组的两个组合不同,至少两个组合中有一个是错误的。 我们测试了与模拟读测试中使用的相同的汇编程序,但使用特定于ont的抛光工具Medaka增加了一个额外的长读抛光步骤。因此,我们生成了未抛光的长只读程序集(使用Miniasm/Minipolish、Raven、Flye和Trycycler)、抛光的长只读程序集(相同的程序集加上Medaka)、长读优先混合程序集(相同的程序集加上Medaka和短读抛光使用Pilon)以及短读优先混合程序集(使用Unicycler)。每种装配方法对每个测试生物体的读集A和读集B都使用。 使用模拟读测试的指标对装配精度进行量化:平均同一性和最差100 bp同一性。这些度量不是基于程序集到引用的对齐(就像模拟读测试所做的那样),而是使用读集a组装的染色体与读集b组装的染色体的对齐。为粘质沙雷氏菌大部分装配方法都无法产生完整的染色体(由于长基因组重复和短的读取N50,参见附加文件)3.: Reads),因此该基因组被排除在外,在分析中留下了6个基因组。与模拟读测试的情况一样,Flye组件在所有抛光阶段的质量都高于Miniasm/Minipolish和Raven组件1:图S3):未打磨(平均身份Q34 vs Q28-Q32;平均最差100-bp同一性81.8% vs 20.2-21.8%), medaka -抛光(平均同一性Q40 vs Q30-Q35;平均最差100 bp同一性94.7% vs 28.2-38.0%)和Medaka+ pilon -polish(平均同一性Q56 vs Q31-Q37;平均最差100 bp同一性94.7% vs 28.2-40.0%)。Flye也是唯一能够为所有6个基因组的两个读集产生完整染色体的长读汇编程序,因此Miniasm/Minipolish和Raven被排除在我们的主要结果之外。 由于在实读测试中,平均恒等和最差100 bp恒等指标无法识别所有装配错误,我们还使用了另外两种方法来评估重新装配的质量。第一个是ALE [33,它使用短读比对组装序列,以产生该组装的可能性得分(得分越高越好),我们将每个基因组标准化,以产生z得分。ALE利用从短读对齐中提取的映射精度、读深度均匀度和插入大小均匀度生成似然评分。第二种全新的评估方法是IDEEL [34,35,它将组装过程中预测蛋白质的长度与已知蛋白质的数据库进行比较。装配中的Indel错误会导致编码序列的帧移导致截断,所以容易出错的装配往往会预测到比最匹配的已知蛋白质更短的蛋白质。我们量化了每个装配中预测的蛋白质的比例,这些比例≥其最佳匹配已知蛋白质长度的95%(分数越高越好)。 数字3.显示了真实读结果:均值同一性,最差100-bp同一性,ALEz和IDEEL全长蛋白质。在平均恒等度量中,Trycycler在所有抛光级别上的表现都优于Flye(抛光前的Q37 vs Q34;Medaka抛光后Q42 vs Q40;在Medaka+Pilon抛光后Q62 vs Q56)。这一优势在最差的100-bp识别指标中也很明显(96.7% vs 81.8%抛光前;Medaka抛光后97.0% vs 94.7%;98.3% vs . Medaka+Pilon抛光后的94.7%)。两种长读优先的杂交方法(Flye+Medaka+Pilon和Trycycler+Medaka+Pilon)都优于Unicycler的短读优先杂交组合(平均Q34和100-bp的最坏同识别率为23.5%)。结果表明:Trycycler组合具有较高的平均ALEz-分数比Flye组件在所有抛光水平(-1.031 vs -1.873抛光前;Medaka抛光后0.419 vs 0.235;Medaka+Pilon抛光后为0.828 vs 0.806)和长读优先混合组装优于unicycle组装(平均ALE)z得分为0.617)。IDEEL的结果显示了同样的趋势,Trycycler装配体比flyye装配体具有更多的全长蛋白(抛光前为78.3% vs 72.3%;93.8%,而Medaka抛光后为91.8%),但所有混合组件在这一指标上表现相当(97.6%)。 图3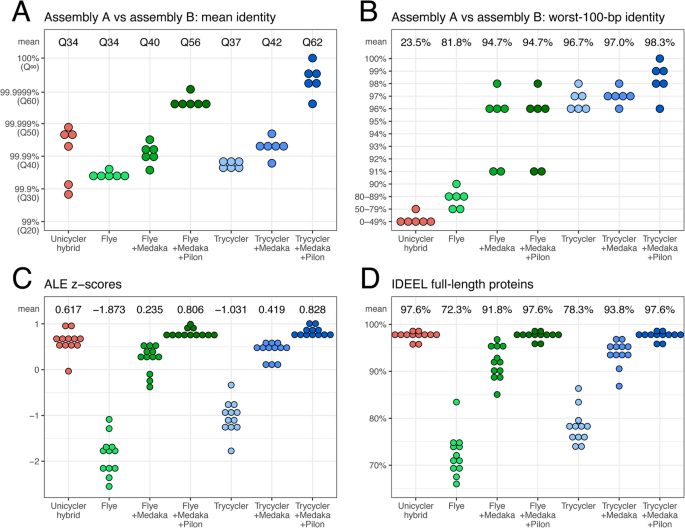
实读测试的结果。对于6个基因组,我们从相同的DNA提取中产生了两个独立的杂交阅读集。然后用Unicycler(短读优先混合程序集)、Flye(长只读程序集)、Flye+Medaka(长只读程序集)、Flye+Medaka+Pilon(长读优先混合程序集)、Trycycler(长只读程序集)、Trycycler+Medaka(长只读程序集)、Trycycler+Medaka+Pilon(长读优先混合组件)。对于每个基因组和每个组装方法,我们将两个独立组装的染色体彼此对齐,以确定平均组装身份(一个)和100个bp滑动窗口中最差的身份(B).对于长时间只读装配,Trycycler始终达到比flyye更高的精度(在Medaka抛光前后)。Trycycler+Medaka+Pilon达到了最高的准确性,比其他混合方法(unicycle和Flye+Medaka+Pilon)做得更好。我们还使用ALE (C)及IDEEL (D).ALE分配一个可能性分数(转换为z-基于每个基因组的分数)到每个组装,根据其与Illumina读取集的一致性。IDEEL在数据库中识别出≥95%与其最匹配的已知蛋白质长度的预测蛋白质的比例 全尺寸图像虽然上面的结果使用了在Trycycler之后的Medaka抛光(即,Trycycler+Medaka),但也可以在Trycycler的输入组件(即,Medaka+Trycycler)或在Trycycler的输入组件和最终组件(即,Medaka+Trycycler+Medaka)上运行Medaka抛光。我们使用实际读取的数据尝试了这些替代方法,发现尽管所有方法的执行情况相似(平均标识为Q41-Q42),但当Medaka是过程的最后一步时获得了最佳结果(附加文件3.:青鳉秩序)。因此,我们推荐Trycycler+Medaka方法,因为它的简单性和准确性。 错误的类型和位置额外的文件1图S4显示了16个基因组(10个模拟基因组和6个真实基因组)装配中的误差位置,并指出了基因组的重复区域。长只读组装(Flye, Flye+Medaka, Trycycler和Trycycler+Medaka)的错误分布在整个基因组中,在重复和非重复序列中都发生。长读优先的杂交组合(Flye+Medaka+Pilon和Trycycler+Medaka+Pilon)通常在重复序列中有较高的错误率,在很多情况下,基因组的非重复序列中没有错误。短读优先的混合程序集(Unicycler)在重复序列和非重复序列中都经常出现错误。Indel错误比替换错误更常见:44%的错误是插入,47%是缺失,9%是替换。对于真正的读取,在Medaka抛光之前,Flye程序集有时会在错误率上出现局部峰值(表示更严重的错误或一组错误),但这些峰值在Medaka抛光之后就不存在了。Trycycler组件没有遇到同样的问题。飞片组件通常在对应于最初的起点/终点的位置上有错误。 在contig的开始/结束处的Flye错误是由不完美的循环引起的:在一个圆形contig的开始/结束处缺失或重复的碱基,我们在之前的长读汇编器基准研究中更详细地描述了这种现象[13].这些错误没有被Medaka或Pilon纠正,因为这些工具没有意识到contig的圆性,也就是说,contig的最后一个碱基应该立即在它的第一个碱基之前。由于我们的分析涉及将所有程序集规范化到一致的起始位置(全局对齐所需),因此在我们的测试中,在contig的开始/结束处丢失/重复的碱基被注册为序列中间indel错误。这些indel误差降低了均值恒等式,如果足够大,也降低了最差的100 bp恒等式。 为了评估循环误差对Flye精度的影响,我们使用原始参考序列(在模拟读测试中)或Trycycler+Medaka+Pilon组件(在实际读测试中)手动固定所有Flye组件的循环。在22个Flye组件中(10个来自模拟读取,12个来自真实读取),4个有完美的循环,5个有重复的碱基,13个有缺失的碱基。最糟糕的Flye循环错误是13 bp的删除,Flye循环错误的平均幅度是3.7 bp(附加文件2和3.: Flye环化)。然后,我们使用Flye组件的固定循环版本重新进行分析,结果显示在附加文件中1:图S5为模拟读取和附加文件1:图S6为真实读取。Flye在这些结果中表现较好,特别是在最差的100-bp恒等度量中,这表明在许多情况下,Flye装配中的圆化误差是最大的单个误差。然而,Trycycler在每个抛光阶段(未抛光、medaka抛光和pilon抛光)仍然比Flye生产更精确的组件。 Trycycler结果的一致性Trycycler不是一个完全自动化的管道——它需要人的判断和干预。这就提出了一个问题:它在不同用户手中的性能如何?为了回答这个问题,我们招募了5名研究人员,他们在生物信息学方面有经验,但没有参与Trycycler的开发。他们获得了用于实际读测试的6个基因组中的每个基因组的ONT读集,并被要求在没有Trycycler开发人员任何帮助的情况下(仅使用Trycycler文档来指导他们)生产Trycycler组件。然后我们比较了产生的组装,观察了contigs的存在/缺失以及染色体序列的一致性(图。4). 图4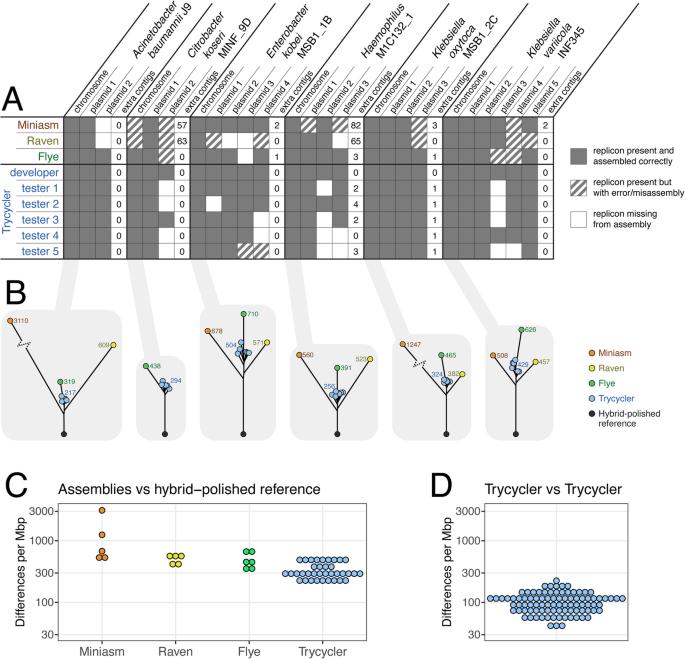
多用户测试的结果,该测试评估了Trycycler组件在不同用户运行时的一致性。结果包括来自三个不同的长读汇编程序(Miniasm/Minipolish、Raven和Flye,对于给定的一组读和参数,它们都是自动的和确定的,也就是说,独立于用户)和来自六个不同用户(Trycycler的开发人员和五个测试人员)的Trycycler汇编程序。一个检测基因组中复制子的存在/缺失矩阵。每个复制子被分类为存在于程序集中、不在程序集中或存在但有错误/装配错误(参见附加文件)4:矩阵获取更多细节)。附加contigs的数量(例如,假序列或污染序列)也表明了每个组装。所有Trycycler装配体都包含一个精确的染色体,只有一个Trycycler装配体包含错误装配。然而,在许多情况下,Trycycler测试人员排除了一个真正的质粒(最常见的是一个小质粒)或包含了一个额外的质粒(最常见的是由跨条码污染读取构建的)。B基于成对排列距离的所有染色体可用集合的邻居连接树。混合抛光(Medaka+Pilon)版本的开发人员的Trycycler组装作为参考序列。这些值表示每个组件和抛光参考之间每Mbp的单bp差异的数量(Trycycler的值是所有六个Trycycler组件的平均值)。对于每个基因组,Trycycler组装紧密聚集,比其他长读组装更接近抛光参考。C每个装配的染色体与杂交抛光参考之间的差异。值为序列每Mbp的单bp差值。平均而言,与单汇编程序集相比,Trycycler汇编程序集包含较少的差异。D每个染色体的Trycycler组合之间的成对差异。值为每Mbp序列的单bp差异,有90个值(每个基因组6个基因组× 15个独特的成对组合)。 全尺寸图像不同用户Trycycler装配体之间差异的主要来源是包含/排除质粒contigs(图。4小的质粒经常对长读汇编器造成问题,这导致它们有时被Trycycler用户排除。Trycycler组件有时包括污染质粒contigs(例如,交叉条形码污染)。复制子有大规模错误或错误组装发生在许多单汇编程序组装(从Miniasm/Minipolish, Raven,和Flye)。这些错误包括分散经由(例如,将一个复制子序列两个叠连群之间),翻一个复制子在一叠连群(例如,组装6-kbp质粒变成12-kbp重叠群),大规模的环化问题(例如,80 kbp的开始/结束重叠),和冗余重叠群(例如,生产五叠连群一个复制子)。这种类型的错误在Trycycler程序集中非常罕见(仅在一种情况下出现)。所有这些错误的详细描述在附加文件中4:矩阵。 为了评估组装序列的一致性,我们建立了一个邻居连接树(基于成对排列距离)的装配染色体为每个基因组(图。4B).将开发商的Trycycler+Medaka+Pilon组件作为参考序列,作为实读测试结果(图2)。3.)表明这些是基因组最准确的代表。对于每个测试分离物,不同用户生成的Trycycler程序集比任何(自动化的)单程序集更接近参考序列(图。4C),不同用户的Trycycler组件之间的差异相对较小(图)。4D).不同用户生成的Trycycler组件之间的所有差异都是小规模的:大多数仅是单bp的差异,最大的差异是串联重复中4 bp的indel(附加文件4: Trycycler vs . Trycycler)。最常见的差异是均聚物序列长度的1-bp差异(占所有Trycycler-vs-Trycycler序列差异的78.5%)。 |
【本文地址】