| GCTA学习6 | 您所在的位置:网站首页 › grm怎么读 › GCTA学习6 |
GCTA学习6
|
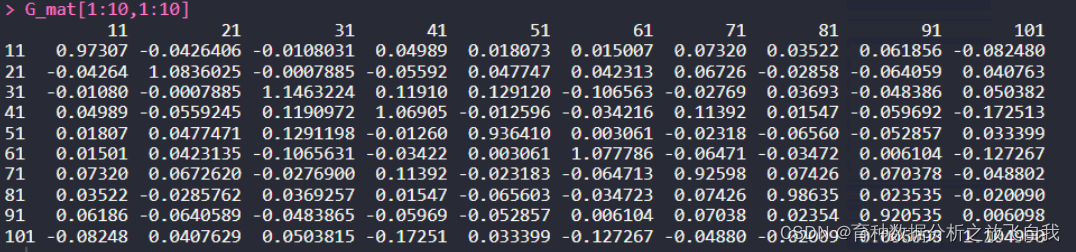
GRM矩阵,全称:genetic relationship matrix (GRM)。 GCTA计算GRM有两种方法 默认的Yang,–make-grm-alg 0Van的方法:–make-grm-alg 1GCTA计算GRM有两种形式 默认的二进制形式:–make-grm,或者 --make-grm-bin文本格式(三元组):–make-grm-gz 1. GCTA计算GRM:二进制下面这两个命令,是等价的。 --make-grmor --make-grm-binEstimate the genetic relationship matrix (GRM) between pairs of individuals from a set of SNPs and save the lower triangle elements of the GRM to binary files, e.g. test.grm.bin, test.grm.N.bin, test.grm.id. 结果会生成矩阵的下三角,保存为二进制文件。 Output file test.grm.bin (it is a binary file which contains the lower triangle elements of the GRM). 这个是二进制存储的GRMtest.grm.N.bin (it is a binary file which contains the number of SNPs used to calculate the GRM). 这个是二进制文件,存储的是参与计算的SNP个数test.grm.id (no header line; columns are family ID and individual ID, see above). 这个是FID和IID的信息。运行命令: gcta64 --bfile ../test --make-grm --make-grm-alg 0 --out g1 2. R函数,读入二进制矩阵可以通过R语言代码读取二进制GRM文件: # R script to read the GRM binary file library(tidyverse) library(data.table) library(ASRgenomics) library(gdata) ReadGRMBin=function(prefix, AllN=F, size=4){ sum_i=function(i){ return(sum(1:i)) } BinFileName=paste(prefix,".grm.bin",sep="") NFileName=paste(prefix,".grm.N.bin",sep="") IDFileName=paste(prefix,".grm.id",sep="") id = read.table(IDFileName) n=dim(id)[1] BinFile=file(BinFileName, "rb"); grm=readBin(BinFile, n=n*(n+1)/2, what=numeric(0), size=size) NFile=file(NFileName, "rb"); if(AllN==T){ N=readBin(NFile, n=n*(n+1)/2, what=numeric(0), size=size) } else N=readBin(NFile, n=1, what=numeric(0), size=size) i=sapply(1:n, sum_i) return(list(diag=grm[i], off=grm[-i], id=id, N=N)) } 3. 将二进制GRM变为N*N的矩阵然后通过下面代码,转换为n*n的G矩阵: aa = ReadGRMBin(prefix = "g1") G_mat = matrix(0,length(aa$diag),length(aa$diag)) diag(G_mat) = aa$diag lowerTriangle(G_mat,byrow = T) = aa$off G_mat = G_mat+t(G_mat)-diag(diag(G_mat)) rownames(G_mat) = colnames(G_mat) = aa$id$V2 G_mat[1:10,1:10]
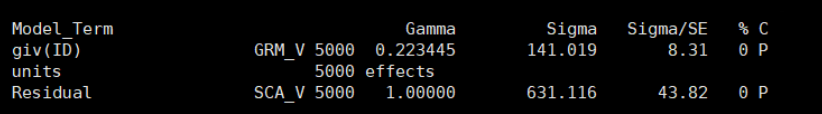
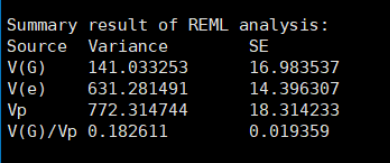
Estimate the GRM, save the lower triangle elements to a compressed text file (e.g. test.grm.gz) and save the IDs in a plain text file (e.g. test.grm.id). 估计的GRM文件,存储矩阵的下三角,压缩文件,存储ID信息 Output file format test.grm.gz (no header line; columns are indices of pairs of individuals (row numbers of the test.grm.id), number of non-missing SNPs and the estimate of genetic relatedness) 生成的文件,为压缩文件,第一列和第二列为编号信息(根据ID的顺序编号,相当于是矩阵的下三角行列信息),第三列是SNP个数,第四列是相关系数 1 1 1000 1.0021 2 1 998 0.0231 2 2 999 0.9998 3 1 1000 -0.0031 ...test.grm.id (no header line; columns are family ID and individual ID) 为FID和IID数据,第一列为家系信息,第二列是个体ID。 011 0101 012 0102 013 0103 ... 5. 将二进制GRM变为ASReml支持的格式ASReml-R的ginv格式,是矩阵的下三角,第一列是矩阵的行号,第二列是矩阵的列号,第三列是矩阵的数值(亲缘关系系数)。所以,可以直接根据GCTA的文本的GRM,进行转换。 注意,ASReml计算需要的是G逆矩阵,而GCTA计算的是G矩阵,所以要求逆矩阵之后,才可以利用。 命令代码: gcta64 --bfile test --make-grm-bin --make-grm-alg 1 --out g1 --maf 0.01在R语言中读取二进制G矩阵,并转化为逆矩阵的三元组形式 ReadGRMBin=function(prefix, AllN=F, size=4){ sum_i=function(i){ return(sum(1:i)) } BinFileName=paste(prefix,".grm.bin",sep="") NFileName=paste(prefix,".grm.N.bin",sep="") IDFileName=paste(prefix,".grm.id",sep="") id = read.table(IDFileName) n=dim(id)[1] BinFile=file(BinFileName, "rb"); grm=readBin(BinFile, n=n*(n+1)/2, what=numeric(0), size=size) NFile=file(NFileName, "rb"); if(AllN==T){ N=readBin(NFile, n=n*(n+1)/2, what=numeric(0), size=size) } else N=readBin(NFile, n=1, what=numeric(0), size=size) i=sapply(1:n, sum_i) return(list(diag=grm[i], off=grm[-i], id=id, N=N)) } aa = ReadGRMBin(prefix = "g1") G_mat = matrix(0,length(aa$diag),length(aa$diag)) diag(G_mat) = aa$diag lowerTriangle(G_mat,byrow = T) = aa$off G_mat = G_mat+t(G_mat)-diag(diag(G_mat)) rownames(G_mat) = colnames(G_mat) = aa$id$V2 #diag(G_mat) = diag(G_mat) + 0.01 ginv = G.inverse(G_mat,sparseform = T)$Ginv head(ginv)然后就可以进行GBLUP评估了:两者结果完全一致。 ASReml的结果: GCTA reml的结果: GCTA学习1 | 抛砖引玉–初步介绍 GCTA学习2 | 软件下载安装–windows和Linux GCTA学习3 | GCTA的两篇NG:fast-LMM和fast-GLMM GCTA学习4 | GCTA说明文档–功能分类及常见问题 GCTA学习5 | GCTA计算PCA及可视化 GCTA学习6 | GCTA计算GRM矩阵(kinship矩阵) GCTA学习7 | 计算单性状遗传力和标准误 GCTA学习8 | GCTA计算多性状遗传力和遗传相关 |
【本文地址】