| circRNA分析工具集 | 您所在的位置:网站首页 › circRNA测序原理 › circRNA分析工具集 |
circRNA分析工具集
|
CIRCexplorer及CIRCexplorer2方法 基本来说算是对方法的翻译,这里只对2进行阐述,如果想更深入的了解还是看原版的好 CIRIexplorer请参考http://yanglab.github.io/CIRCexplorer/ CIRIexplorer2请参考http://circexplorer2.readthedocs.io/en/latest/ CIRCexplorer2是一个全面综合的circRNA分析工具集,是CIRCexplorer的延续。 作者:Xiao-Ou Zhang([email protected]), LiYang([email protected]) 维护:Xu-Kai Ma([email protected]) 教程主要包括原理及流程、软件安装及数据准备、circRNA的识别与注释、从头组装新的circRNA、circRNA的可变剪切,另外还提供了一步流程。 一、原理及流程 主要包括两部分,一个是注释流程,一个是描述流程 注释流程
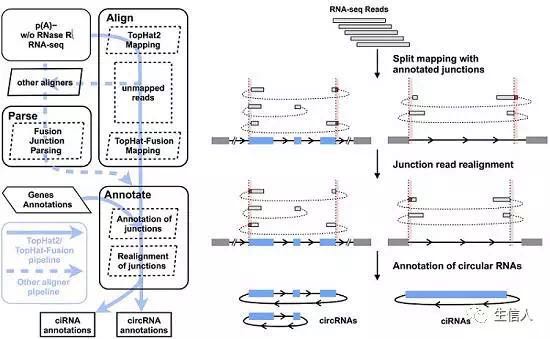
该流程与CIRCexplorer一致,是一个整合策略来识别fusion junction reads(可能来自与反向剪切的外显子或来自内含子套索)。CIRCexplorer2的特征是可以支持多种比对工具。 对circRNA稍有了解的从图中就能看出CIRCexplorer的基本思想。该方法主要依赖与基因的注释,实际上是利用已注释的外显子剪接边界(不知道还有没有其他类型的边界,我的理解是外显子与内含子的界限)来识别具有高准确性的circRNA,作者也提供了识别不精确边界的方法。流程图左右两个面板,其实右边的面板更直观一些,包括数据的输入、双末端比对到外显子边界(红色的表示fusion junction)、候选junction的再比对(即将之前的fusion进行分割再比对)以及注释。 该流程主要包含两步,会详细地在后边阐述 1. Alignment and Parsing 2. Annotating 描述流程
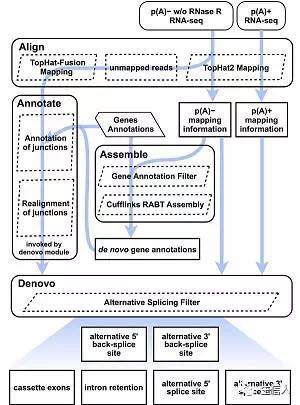
该流程全面而系统地描述了circRNA的可变剪切事件,主要通过de novo组装的方式 1. 整个流程看着比较复杂,实际上使用了Cufflinks的RABT方法。 2. 除了已注释的外显子,该方法还能识别到circRNA特异的外显子,这些外显子并不在线性RNA中表达。 3. 该方法可以识别2中可变反向剪切事件(5'和3'的剪接位点的改变)以及四种可变剪切事件(与线性转录本差不多,不知道的童鞋请访问WikiPedia 该流程主要包括3步,后面会详细介绍 1. Alignment 2. Assembly 3. Alternative Splicing 二、 软件安装及数据准备 由于我只使用过TopHat,所以这里只介绍这个了。以下是软件需求,python相关软件包都被集成到了CIRCexplorer2中,所以也不用费事儿预先安装了(pandas需要自己装)。 软件 TopHat(>=2.0.9) Cufflinks(>=2.1.1) BEDTools UCSC Utilities genePredToGtf gtfToGenePred o python(2.7+) pysam(>=0.8.4) pybedtools(require pandas if pybedtools>=0.7.6) docopt scipy(only unsed in denovo module) 小编研究过程中,由于没有权限安装根目录软件。经过长期的洗礼,现在也大概知道咋捣鼓了。其他的软件都比较好安装,就是python不太听使唤,pip命令如果不好使,一般我就用github上的开发版。 CIRCexplorer2安装 (1) pip CIRCexplorer2已经发布到了PyPI,如果Python是安装在自己的路径下,请设置pip的环境变量——将~/python/lib/python2.7/site-packages添加进~/.bashrc。另外,这个方法会自动安装python的模块pysam/pybedtools/docopt等。 pip install circexplorer2 (2) source 该方法发布到了github上,所以可以用git的方式安装 git clone https://github.com/YangLab/CIRCexplorer2.git cd CIRCexplorer2 pip install -r requirements.txt ### install scipy according to http://www.scipy.org/install.html python setup.py install conda 这是啥我就不太清楚了,请自己查询 数据准备 (1)RNA-seq 推荐使用poly(A)-的ribo- RNA-seq数据(即rRNA删除的数据),另外ribo- tatal RNA也适用。对于一些ribo+的数据需要自己先处理,不过大部分原始公共数据都删除了rRNA。当然RNase R处理的数据能够富集circRNAs。 (2)参考基因组fasta文件 这个可以直接从UCSC、Ensembl或者GENCODE下载,既有整合在一起的染色体参考基因组fasta,也有单个染色体的fasta。基因注释文件 该文件的格式是UCSC GenePred格式的扩展,主要包括以下字段:
该文件的获取你完全不用担心,作者提供了相应的程序来获得注释文件,包含RefSeq/KnownGenes/Ensembl注释以及人类和小鼠的基因组(fa) fetch_ucsc.py hg19/hg38/mm9/mm10 ref/kg/ens/fa out 例如 下载Ensembl基因注释和hg19参考基因组 fetch_ucsc.py hg19 ens hg19_ens.txt fetch_ucsc.py hg19 fa hg19.fa 实际上fetch_ucsc.py连接的是UCSC数据库中的文件,但UCSC中没有构建Ensembl hg38的注释文件(让人很不爽),所以喜欢用Ensembl的童鞋需要自己构建注释文件。首先利用gtfTOGenePred将Ensembl的注释文件转化为UCSC GenePred, 然后提取gtf中基因symbol与转录本id的对应关系,最后merge两个文件就能得到CIRCexplorer2的标准格式。 另外,注释文件也能转化为gtf文件 cut -f2-11 hg19_ens.txt | genePredToGtf file stdin hg19_ens.gtf 当然,将3个数据库注释文件整合作为CIRCexplorer2输入也是一个不错的选择。 cat hg19_ref.txt hg19_kg.txt hg19_ens.txt > hg19_ref_all.txt build Bowtie与Bowtie2 index 使用TopHat的童鞋一定不要忘了要先构建index,当然,CRICexplorer2也提供了不构建index的方法。 对于index的关联,我不太清楚具体,Bowtie的帮助文档和网友说可以添加export BOWTIE_INDEXES=[index path]关联,但是我没有实现这种方式;最直接的方式就是将index构建在与参考基因组相同的目录下,并且名字一样。完全不用担心bowtie与bowtie2 index后缀,它们不一致。 bowtie-build hg19.fa hg19 bowtie2-build hg19.fa hg19 三、 circRNA的识别与注释 完事具备,终于要开工了。前面大家也看到了,CIRCexplorer2实际上和其他方法差不多,就是想找fusion junction(每个人的称谓不一样,有人称为scrambled read。Whatever!)。似乎CIRCexplorer主要是处理single-end测序数据的,所以原教程主要以单末端形式进行。似乎双末端数据也能转化单末端的形式,但这里还是以双末端方式进行。在我的印象里,双末端数据(主要是Illimina)数据才是主流吧。CIRCexplorer2加入了豪华套餐,能够对双末端进行处理,但只是针对Tophat比对工具。 该流程主要包括3部分:Align/Parse/Annotate。 • Align tophat2 -o [tophat_fusion] -p 15 --fusion-search --keep-fasta-order --bowtie1 --no-coverage-search \ [bowtie1_index] [RNAseq_1.fastq] [RNAseq_2.fastq] o 输入 -p 15指定15个CPU内核来运行该程序; --keep-fasta-order --bowtie1 --no-coverage-search感兴趣的童鞋可以查看Tophat的帮助文档; -o [tophat_fusion]中[tophat_fusion]为结果输出文件夹; [bowtie1_index]为bowtie的index的名字(bowtie index后缀为.ebwt),之前也提到了最好将index与基因组fasta放在同一个目录,名字也一致; [RNAseq_1.fastq]为双末端数据的第一个文件。 o 输出 结果将会输出在[tophat_fusion]文件夹中,包含许多日志文件,以及比对上的fusion reads包含在accepted_hits.bam中——这也是下一步的输入。 • Parse CIRCexplorer2 parse --pe -t TopHat-Fusion [tophat_fusion/accepted_hits.bam] -o [circ_out] > CIRCexplorer2_parse.log o 输入 --pe表示使用的是双末端方法; [tophat_fusion/accepted_hits.bam]就是Align的输出。 o 输出 -o [circ_out]为输出文件夹,最后会得到一个fusion_junction.bed文件; CIRCexplorer2_parse.log为该过程的日志文件。 Annotate ``` CIRCexplorer2 annotate -r REF -g GENOME [circ_dir] > CIRCexplorer2_annotate.log ``` 令人不爽的是,该命令行不能使用\多行书写,这使代码显得非常ugly!这一步实际上就是根据已有的外显子边界,将fusion junction reads精确地注释上去。 o 输入 -r REF这里需要输入的就是之前用fetch_ucsc.py获得的注释文件,例如hg19_ens.txt; -g GENOME当然就是输入参考基因组fasta了; [circ_dir]第二步Parse中的输出文件夹; CIRCexplorer2_annotate.log日志文件 另外还有两个参数可供使用: --no-fix对于那些具有较少基因注释的物种有用 --low-confidence能够提取低置信的circRNA o 输出 很遗憾没有提供输出文件位置的参数。最终会在[circ_dir]中生成文件夹[annotate],其中包含两个文件 annotated_fusion.txt:包含已注释的fusion junction信息,字段信息原说明文档没给,自己猜吧~,或者问作者; circ_fusion.txt:包含circRNA的注释信息,包括的字段如下,前十二列为bed格式: |
【本文地址】