| STAR | 您所在的位置:网站首页 › NM0043336外显子15 › STAR |
STAR
|
1. 将reads通过STAR比对到参考基因组,筛选出Junction reads(1条read含有两个基因融合断点的read )和Spanning reads (R1,R2比对到不同基因上的reads)作为候选融合基因序列。 2. 将候选融合基因与参考基因组注释文件比对,再根据overlap预测融合基因。根据最少reads支持准则以及利用校验工具FusionInspector进行进一步的矫正分析的结果。 3. 过滤预测结果,去除假阳性。 2.1 前期准备 除了二代测序reads,分析前还需要准备好CTAT的 人参考基因组数据库比对文件(也可根据物种生成比对数据库)。由于数据库一直在更新,数据库版本需和STAR软件的版本匹配。
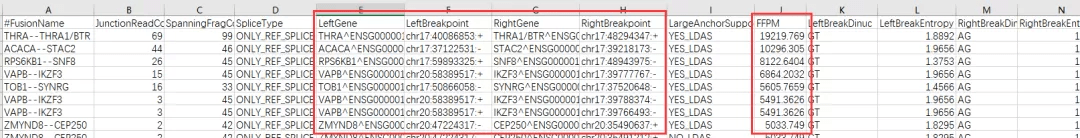
图2 参考基因组信息 2.2 从FASTQ文件开始STAR-Fusion 准备好基因组数据库和二代测序文件后,我们便可以通过STAR-Fusion,来进行融合基因的预测(策略1)。 分析中通过STAR,将reads比对到基因组,并通过各项硬指标筛选获取最合适的分析结果,例如剔除MT信息,限制FFPM最小值,剪接异构体分为多个条目等。输出结果为一个表格文件,示例如下:
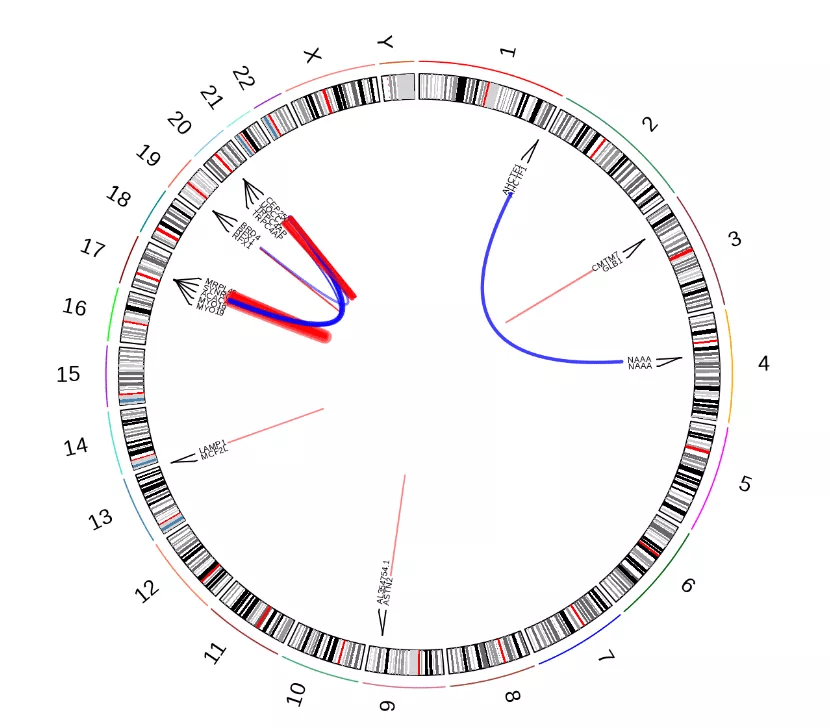
图3 融合基因分析结果 StarFusion 结果说明: FusionName:融合基因名称; JunctionReadsCount:包含在假定的融合连接位点处,一条read可以拆分匹配到两侧融合基因的reads数目; SpanningFragsCount:包含融合连接的reads数目,reads片段的R1,R2两端对应的基因不同; SpliceType:融合基因断点位置是否出现在参考转录本结构注释(例如gencode)所提供的参考外显子连接处; Left/RightGene:融合基因左侧/右侧基因; Left/RightBreakpoint:融合基因断点左/右侧染色体位置信息; LargeAnchorSupport:在假定断点的两侧是否有reads的较长碱基序列(>=25bp)匹配,缺乏LargeAnchorSupport的融合基因通常是假阳性。 FFPM:支持融合的reads的标准化结果,即每百万总reads数的融合量; Left/RightBreakEntropy:位于断点两侧的15个外显子碱基的“香农熵”。最大熵为2,最低为零,低熵位结果通常应被认为低可信度。2.3 circlized 绘制圈图 结果表格内容太多,如何更直观的查看融合基因的分布呢?这里,我们可以使用R软件绘制染色体融合基因的circos图,其中红色连线指同一染色体的基因融合;蓝色连线指不同染色体的基因融合。
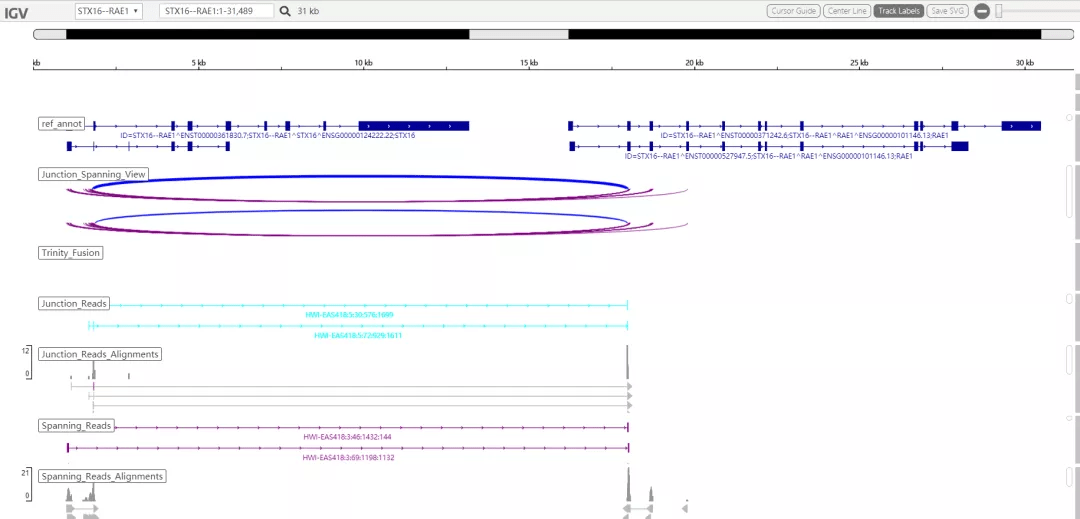
图4 融合基因 染色体圈形图 3、进阶分析分析结果这么多当然不够,我们还需要通过PCR、qPCR来进一步验证融合基因/融合序列在样本中的表达情况,那么如何获取融合序列呢? 3.1 FusionInspector 融合序列可视化 FusionInspector是STAR-Fusion高级分析的第一步,通过对融合基因的序列进行监督分析,拼接比对,进而协助分析人员发现可信度更高的融合转录本。 FusionInspector分析首先会提取融合基因的基因组信息,构建新的候选融合基因组;然后将测序reads与候选融合基因组比对,识别融合断点处的覆盖情况。分析结果有很多,我们只需将以下文件导入IGV软件,即可进行可视化分析。文件如下, 1. finspector.fa:候选融合基因组序列 2. finspector.gtf:候选融合基因组注释文件 3. junction.reads.bam & spanning.reads.bam:候选融合reads比对结果 以下是FusionInspector分析结果的IGV展示。可以看到,融合位点的覆盖情况,融合基因的左/右侧信息都展示在图中。JunctionReads可以完整覆盖融合位点的左右侧,SpanningReads则是不同方向reads覆盖不同的基因。
图5 融合基因 IGV 可视化结果 3.2 Trinity重建融合转录本 获得候选基因组后,就可以重新构建融合转录本。Trinity软件将比对上候选融合基因组的reads重新组装,获得候选融合转录本,然后再使用gmap软件把候选转录本与候选融合基因组进行比对,优化候选转录本的位置信息,提取出最合适的融合转录本(策略2)。得到的结果主要包括以下文件, 1. finspector.gmap_trinity_GG.fusions.fasta:融合转录本序列fa文件 2. finspector.gmap_trinity_GG.fusions.gff3:融合转录本gff3注释文件 这样,我们就得到了一套完整的融合基因分析结果,以便进行后续的分析和验证 1. 融合基因位点信息; 2. 融合基因/转录本序列信息和注释信息; 3. 融合基因可视化结果。 当然,我们也可以从fasta中选取一条序列,通过NCBI进行blast比对。 示例结果我觉得很OK,您意下如何?
图6 融合转录本比对结果示例 目前,ATLAS数据库[2]已经收录了一万多个融合基因位点,除了部分白血病相关的融合基因,其它融合基因在肿瘤发生过程中的功能和作用,我们知之甚少。基因的融合机理、融合基因是否可做为潜在的药物靶点、相关疾病的检测、治疗以及预后,作为医学和生命科学研究方向的热点,具有很大的发展潜力。 参考文献1. Haas B J , Dobin A , Li B , et al. Accuracy assessment of fusion transcript detection via read-mapping and de novo fusion transcript assembly-based methods[J]. Genome Biology, 2019, 20(1):1-16. 2. http://atlasgeneticsoncology.org/返回搜狐,查看更多 |

【本文地址】