零基础入门转录组分析 |
您所在的位置:网站首页 › 转录组数据分析软件有哪些 › 零基础入门转录组分析 |
零基础入门转录组分析
|
零基础入门转录组分析——第三章(质控及数据过滤)
目录
零基础入门转录组分析——第三章(质控及数据过滤)1. 查看原始数据2. 质控3. 质控进阶4. 质控结果合并5. multiqc结果解读(1)General Statistics(2)Sequence Counts(3)Sequence Quality Histograms(4)Per Sequence Quality Scores(5)Per Base Sequence Content(6)Per Sequence GC Content(7)Per Base N Content(8)Sequence Length Distribution(9)Sequence Duplication Levels(10)Overrepresented sequences和Adapter Content
6. 数据过滤
(我这里使用的虚拟机是vmwarewokstation,版本16.0.0, linux系统是ubantu64位,版本20.04.3)
上一章我们准备了原始数据和参考基因组以及注释信息,这一章我们要用到上一章准备好的原始数据。
本实验选用的是模式生物——C57BL/6J小鼠。
实验分组:药物处理组,对照组,每组6只鼠。
软件:vim,fastqc,multiqc, trim-galore
1. 查看原始数据
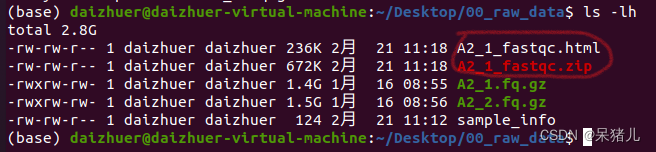
上一章我们在linux桌面上创建了个名为00_raw_data文件夹,并将所有原始数据拷贝到了文件夹中,接下来的操作需要通过终端指令完成。 (1)通过cd Desktop/00_raw_data/切换到原始数据文件下。 (2)输入ls -lh查看当前文件夹下所有文件的详细信息,如下图所示。 (1)保存好样本信息后,接下来做质控,回到终端,输入:fastqc A2_1.fq.gz,软件就会开始对样本做质量分析,分析过程如下图所示。 (2)运行结束后当前目录下会出现两个文件:一个是html文件,一个是zip压缩包,这里我们重点关注html文件(因为html文件里存有质控的结果信息)。 刚才我们通过fastqc A2_1.fq.gz指令输出了一个样本的质控信息,但是我们一共有12个样本,为了节省时间,需要写一个脚本让系统自动运行。(这一步就会用到第一步准备好的样本信息表) (1)第一步创建的sample_info文件中第9列是所有样本的名称。 (2)输入指令:awk '{print "fastqc "$9}' sample_info > fastqc_run.sh这段指令的意思:仅提取sample_info文件第9列,并在每一行前面加上fastqc,最后输出结果为fastqc_run.sh脚本。 (3)cat fastqc_run.sh查看脚本,如下图所示每一行都是: fastqc XXX.fq.gz
(4)最后通过bash fastqc_run.sh指令执行脚本,这时系统就开始一行一行执行代码,直到最后一个样本被分析完。 注:脚本执行后应该会产生大量的html文件和zip文件,如果感兴趣的小伙伴可以一一通过生成的html文件查看每个样本的质量分析结果。 4. 质控结果合并通过指令multiqc .对当前目录下所有质控结果合并,正常运行过程如下图所示。
理想情况应该是左侧非常高,往右越低越好,趋于直线,如上图,就代表序列相对重复较高。 (10)Overrepresented sequences和Adapter Content
注:最后这两个值一般都没什么问题,但是接头含量要注意,下一步质量过滤的时候会去除接头。 从上图我们可以看到除了重复序列,其他的指标基本都没问题,实际上转录组数据一般不看重复序列,因为 这个很有可能是由于表达量高造成的多条重复序列,一般情况可以看看比对效率,GC含量,多比对结果这些,如果 没问题就可以。 6. 数据过滤如果我们拿到的原始数据含有接头或数据质量不好,就需要先进行数据过滤后才能进行后续分析。 数据过滤要用到的软件是trim-galore 首先,我们要了解trim-galore的参数(我这里只介绍部分会用到的参数,如果对全部参数感兴趣,可以自行百度或在linux终端下通过trim_galore -help指令查看。) --quality:设定Phred quality score阈值,默认为20,表示质量分数小于20的会被舍弃,分析时可改成25,稍微严格一些。 --stringency:设定可以忍受的前后adapter重叠的碱基数,默认为1(非常苛刻),值越大越宽松,可以根据实验需求适度放宽。 --length:设定输出reads长度阈值,就是长度不达标的reads会被抛弃。 --paired:对于双端测序结果,一对reads中,如果有一个被剔除,那么另一个会被同样抛弃,而不管是否达到标准。 --output_dir:输出目录,就是过滤后的文件输出的路径(需要提前建立目录,否则运行会报错)。在linux系统桌面下首先要通过指令mkdir 02_clean_data创建一个文件夹名为02_clean_data(需要提前创建好输出文件夹,否则会报错),之后通过cd 00_raw_data切换到存放原始数据的文件夹目录下,开始样本质量过滤 质量过滤代码如下: trim_galore --quality 25 --stringency 1 --length 50 --paired --output_dir /home/daizhuer/Desktop/02_clean_data/ A2_1.fq.gz A2_2.fq.gz我这里设定的quality是25,代表质量小于25会被剔除;stringency参数是1,代表对接头种重叠碱基数检查非常严格,如果接头中重叠碱基数大于1在后面的分析报告中就会显示出来;length参数我设定的是50,表明reads长度小于50的会被弃去;paired表明是双端测序;output_dir输出路径是桌面上的02_clean_data文件夹,质量过滤的结果会保存到这个文件夹中,最后两个是传入的原始文件,因为是双端测序,所以需要上传两个文件。 注:这一步比较慢,运行结束后,02_clean_data文件夹下会生成四个文件,XXX.fq.gz是过滤后的文件,XXX.txt是质量过滤的结果报告,打开XXX.txt文件,如下图所示是质量过滤结果的详细报告。 结语: 以上就是质控及数据过滤的所有过程,如果有什么需要补充或不懂的地方,大家可以私聊我或者在下方评论。 如果觉得本教程对你有所帮助,点赞关注不迷路!!! 目录部分跳转链接:零基础入门生信数据分析——导读 |
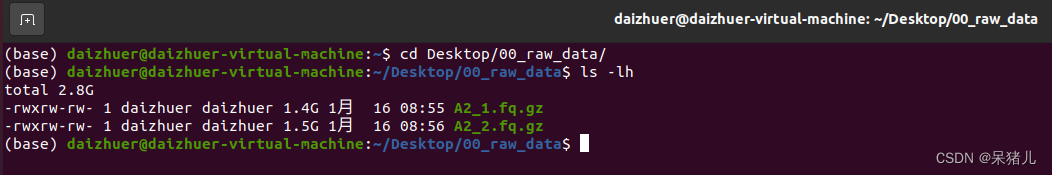 注:为了方便后续的显示和操作,我这里只拷贝了两个原始文件。 (3)准备样本信息文件,输入指令:ls -lh > sample_info,这时你能发现会多出来一个sample_info的文件。 (4)输入:vim sample_info打开文件,文件信息如下图所示。
注:为了方便后续的显示和操作,我这里只拷贝了两个原始文件。 (3)准备样本信息文件,输入指令:ls -lh > sample_info,这时你能发现会多出来一个sample_info的文件。 (4)输入:vim sample_info打开文件,文件信息如下图所示。 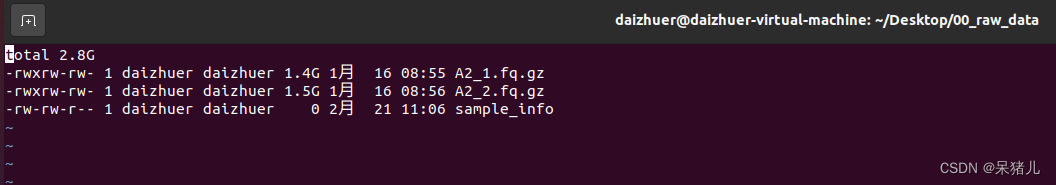 (5)键盘上点一下字母i(为了开始编辑模式),删除第一行和最后一行的内容,处理后的数据如下图所示一共是9列。
(5)键盘上点一下字母i(为了开始编辑模式),删除第一行和最后一行的内容,处理后的数据如下图所示一共是9列。 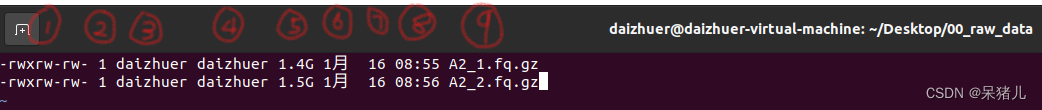 (6)处理后,先点一下键盘上Esc,之后输入:wq即可保存退出。
(6)处理后,先点一下键盘上Esc,之后输入:wq即可保存退出。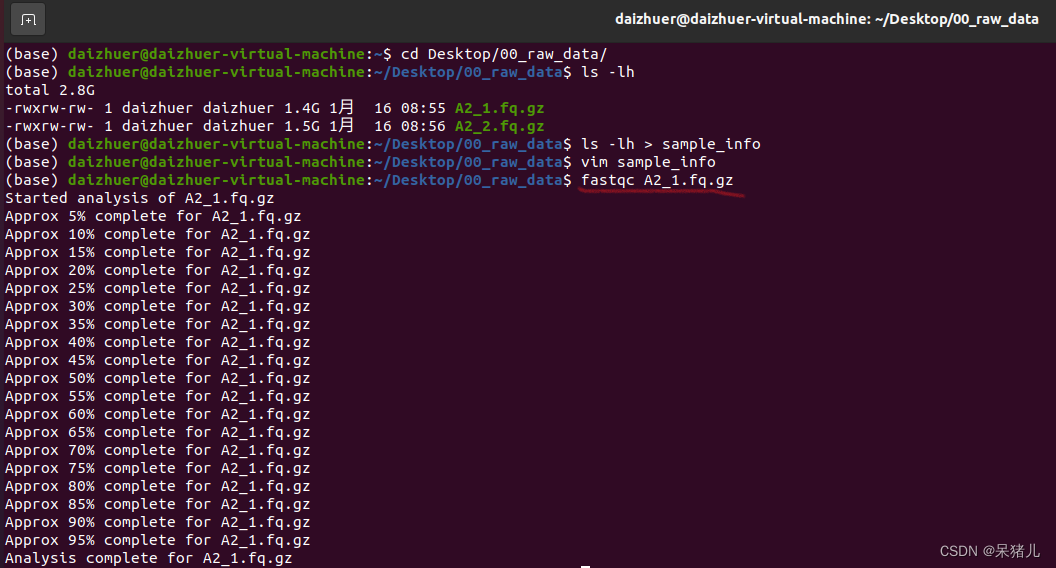
 (3)打开linux左侧收藏栏中文件资源管理器
(3)打开linux左侧收藏栏中文件资源管理器  (4)逐层找到00_raw_dara文件夹中的html文件,双击就可打开查看质控结果(能打开是因为ubantu自带火狐浏览器,也可以拖拽到宿主机中再查看)
(4)逐层找到00_raw_dara文件夹中的html文件,双击就可打开查看质控结果(能打开是因为ubantu自带火狐浏览器,也可以拖拽到宿主机中再查看) 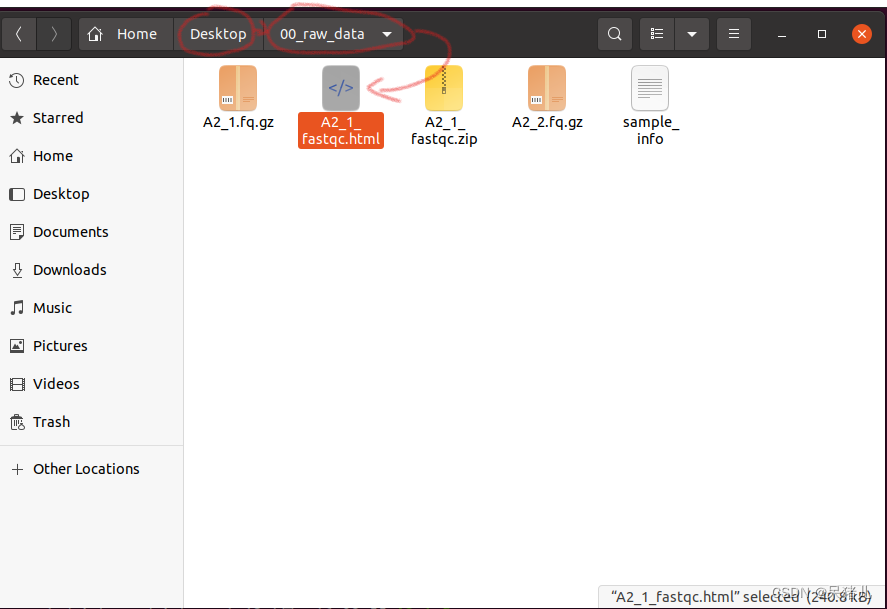 注:质控结果的分析可以参考质控结果分析1,质控结果分析2。
注:质控结果的分析可以参考质控结果分析1,质控结果分析2。
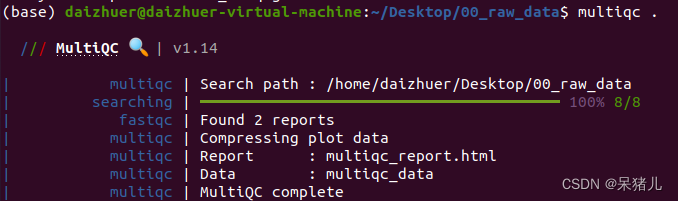 指令运行结束后会在当前目录下输出一个名为multiqc_data的文件夹和一个名为multiqc_report.html的网页,重点关注multiqc_report.html文件,查看方式同步骤二。
指令运行结束后会在当前目录下输出一个名为multiqc_data的文件夹和一个名为multiqc_report.html的网页,重点关注multiqc_report.html文件,查看方式同步骤二。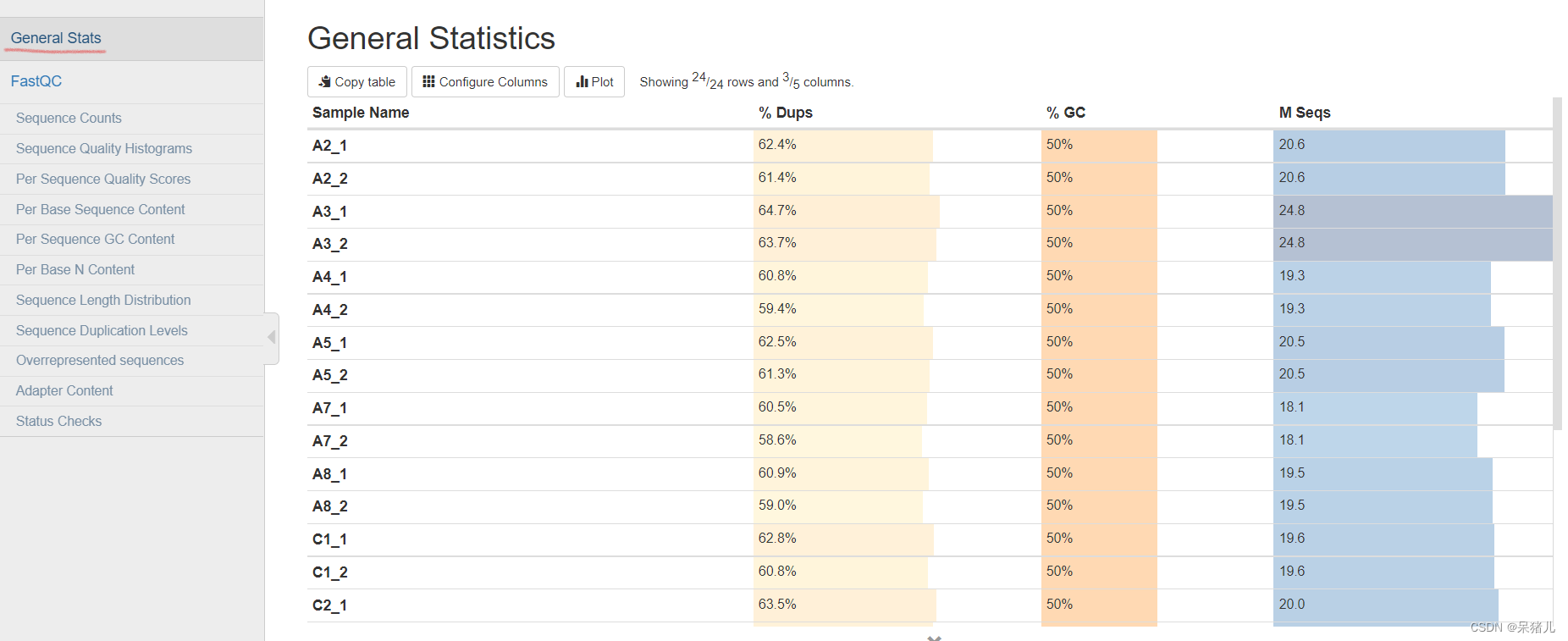 General Statistics——所有样本数据基本情况统计 %Dups——重复reads的比例,比例越高代表重复reads数量就越多,有用的reads就少。 %GC——GC含量占总碱基的比例,比例越小越好 M Seqs——总测序量(单位:millions)
General Statistics——所有样本数据基本情况统计 %Dups——重复reads的比例,比例越高代表重复reads数量就越多,有用的reads就少。 %GC——GC含量占总碱基的比例,比例越小越好 M Seqs——总测序量(单位:millions)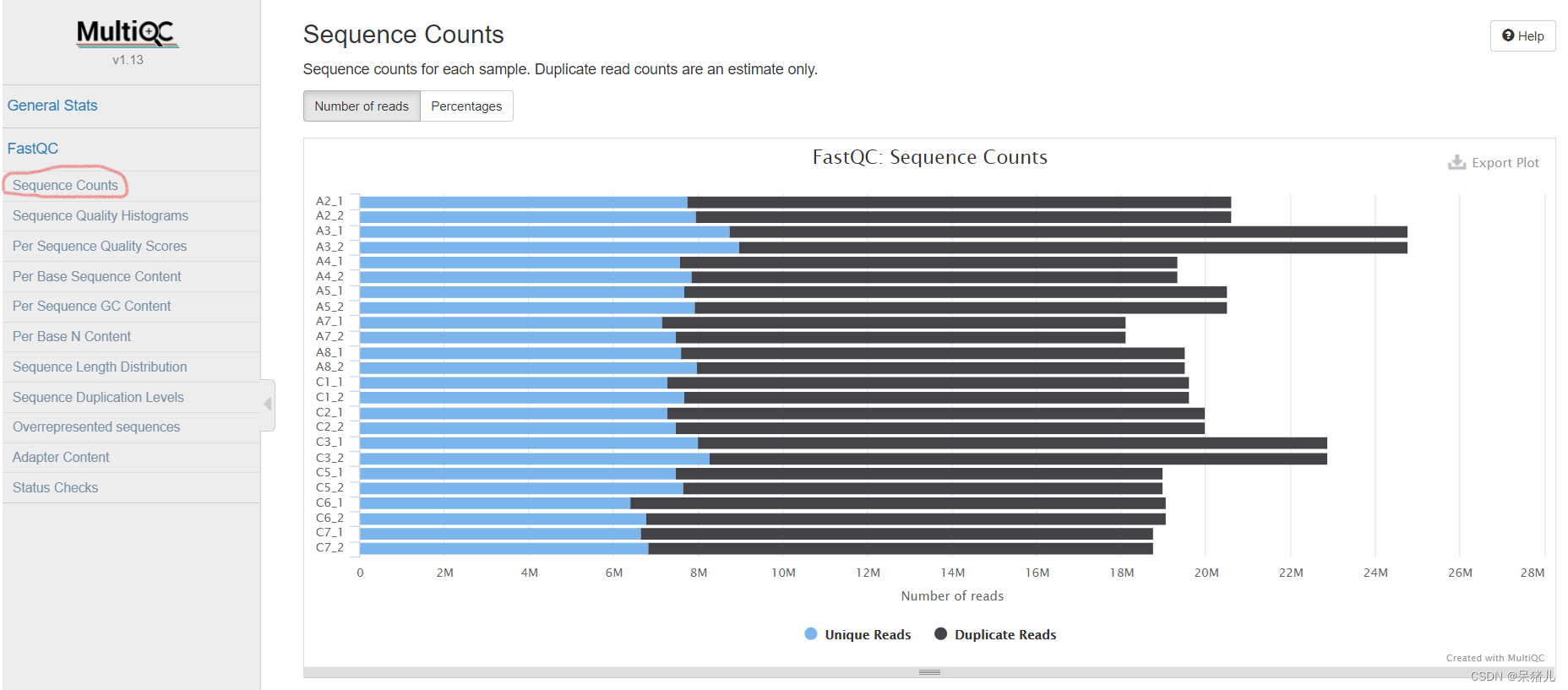 这里可以每个样本重复reads比例,鼠标移到每一个柱子上时会显示每个样本的比例,黑色代表重复的reads,蓝色表示没有重复的reads。
这里可以每个样本重复reads比例,鼠标移到每一个柱子上时会显示每个样本的比例,黑色代表重复的reads,蓝色表示没有重复的reads。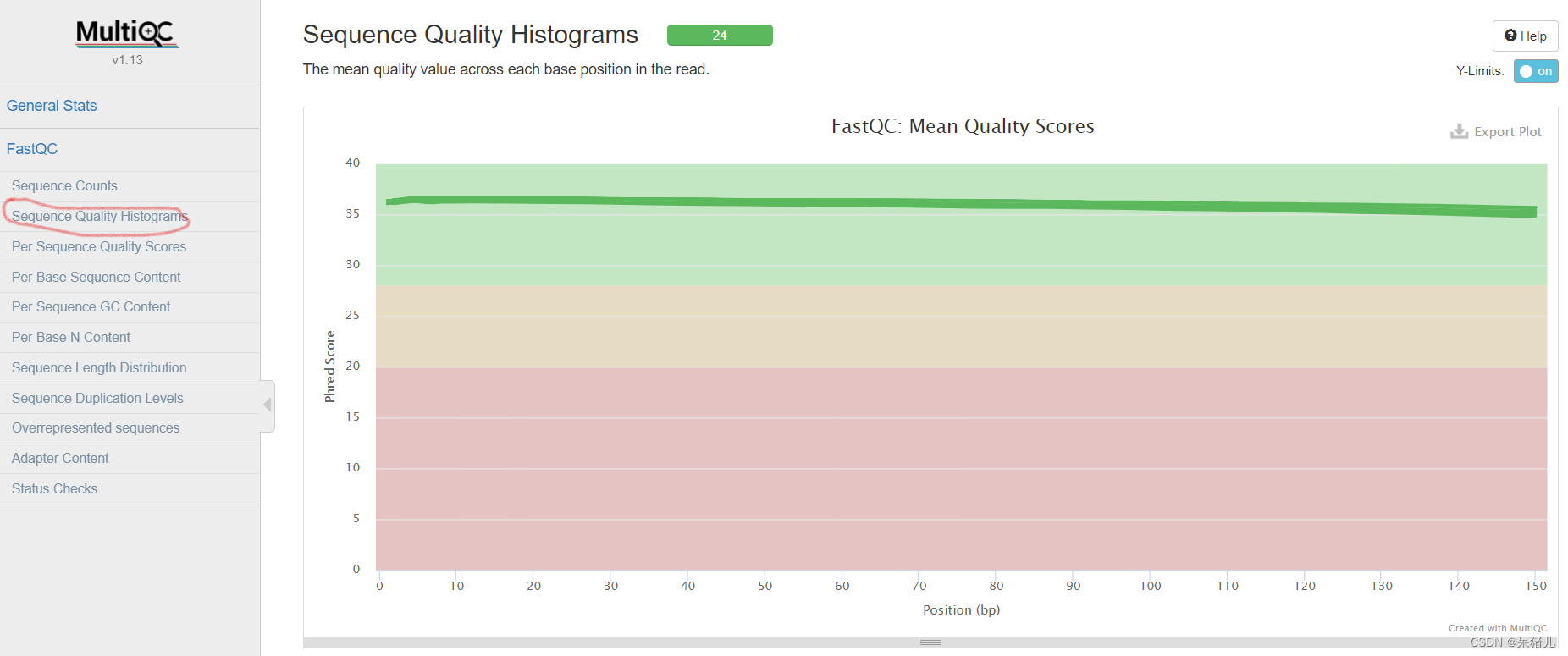 每个read各位置碱基的平均测序质量
每个read各位置碱基的平均测序质量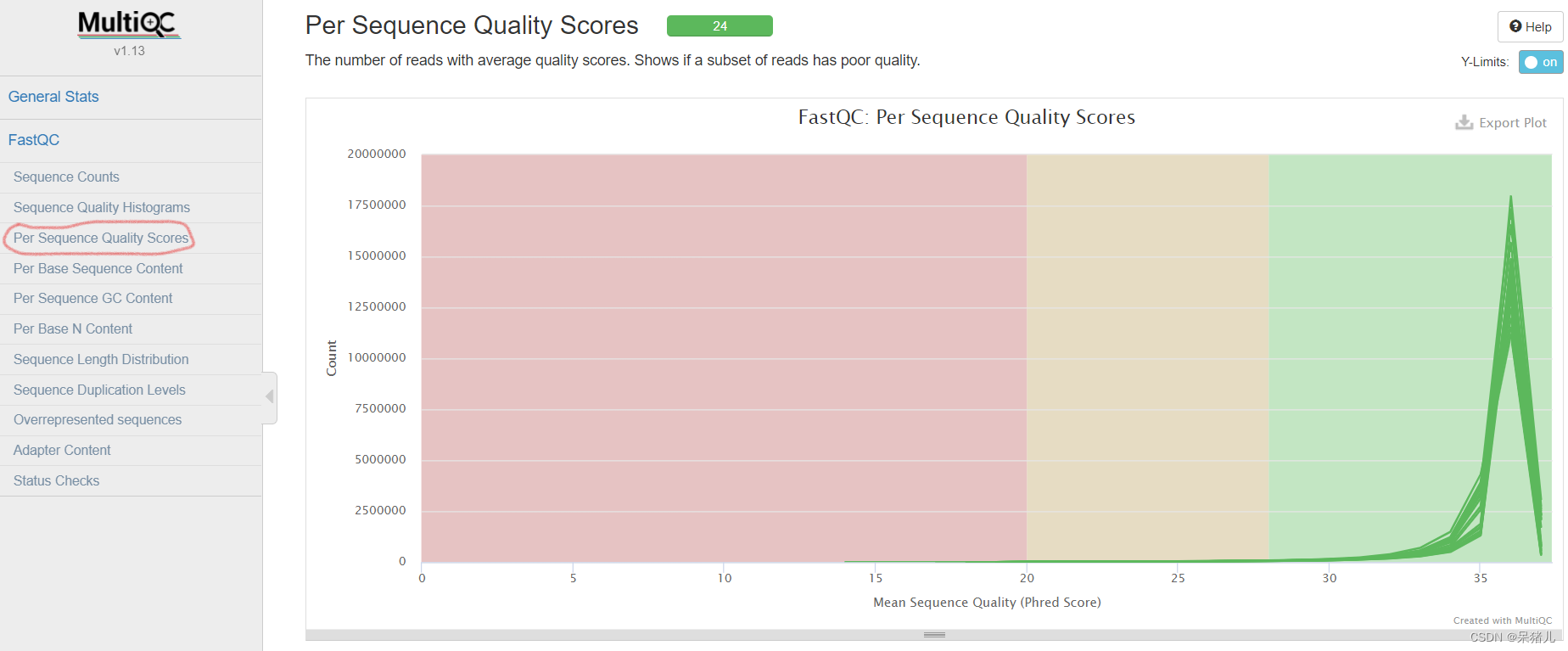 具有平均质量分数的reads的数量
具有平均质量分数的reads的数量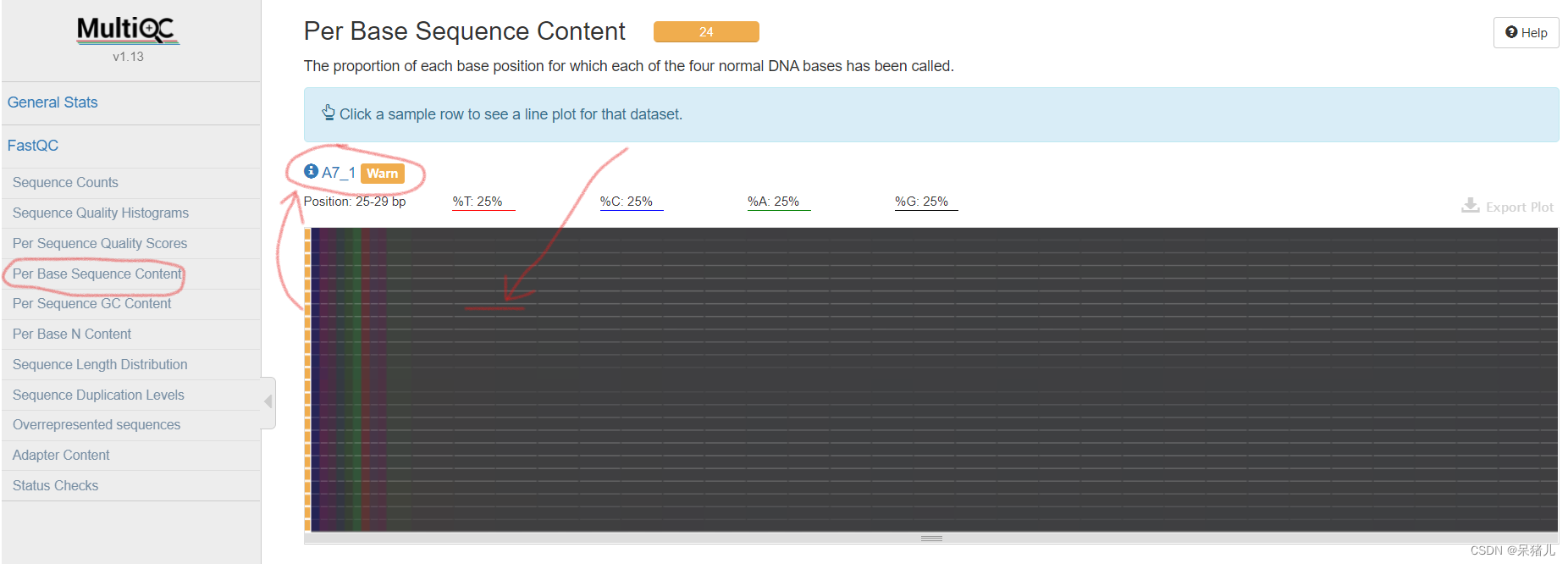 表示每个样本各位置碱基ATCG的比例,一般前一段ATCG比例会有波动,属于正常情况
表示每个样本各位置碱基ATCG的比例,一般前一段ATCG比例会有波动,属于正常情况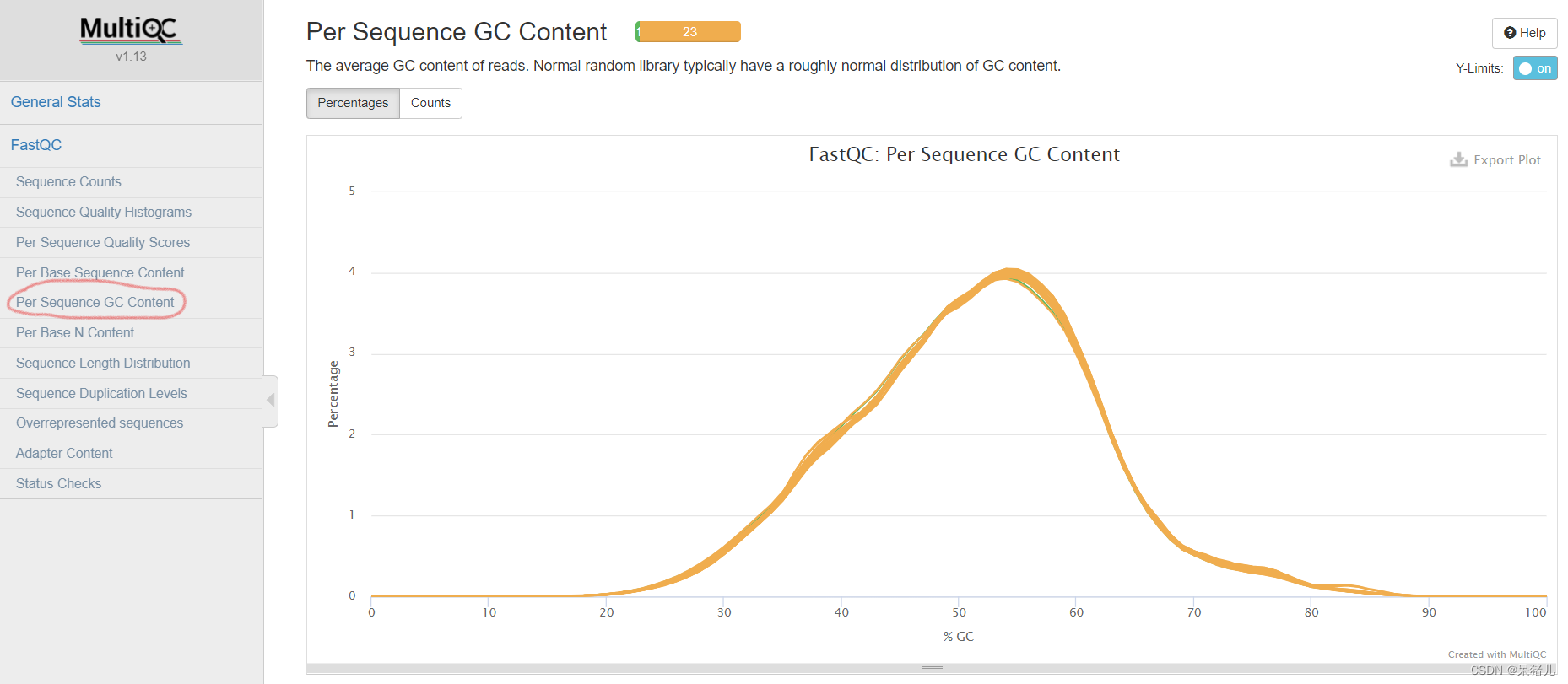 reads的平均GC含量 正常的样本的GC含量曲线会趋近于正态分布曲线
reads的平均GC含量 正常的样本的GC含量曲线会趋近于正态分布曲线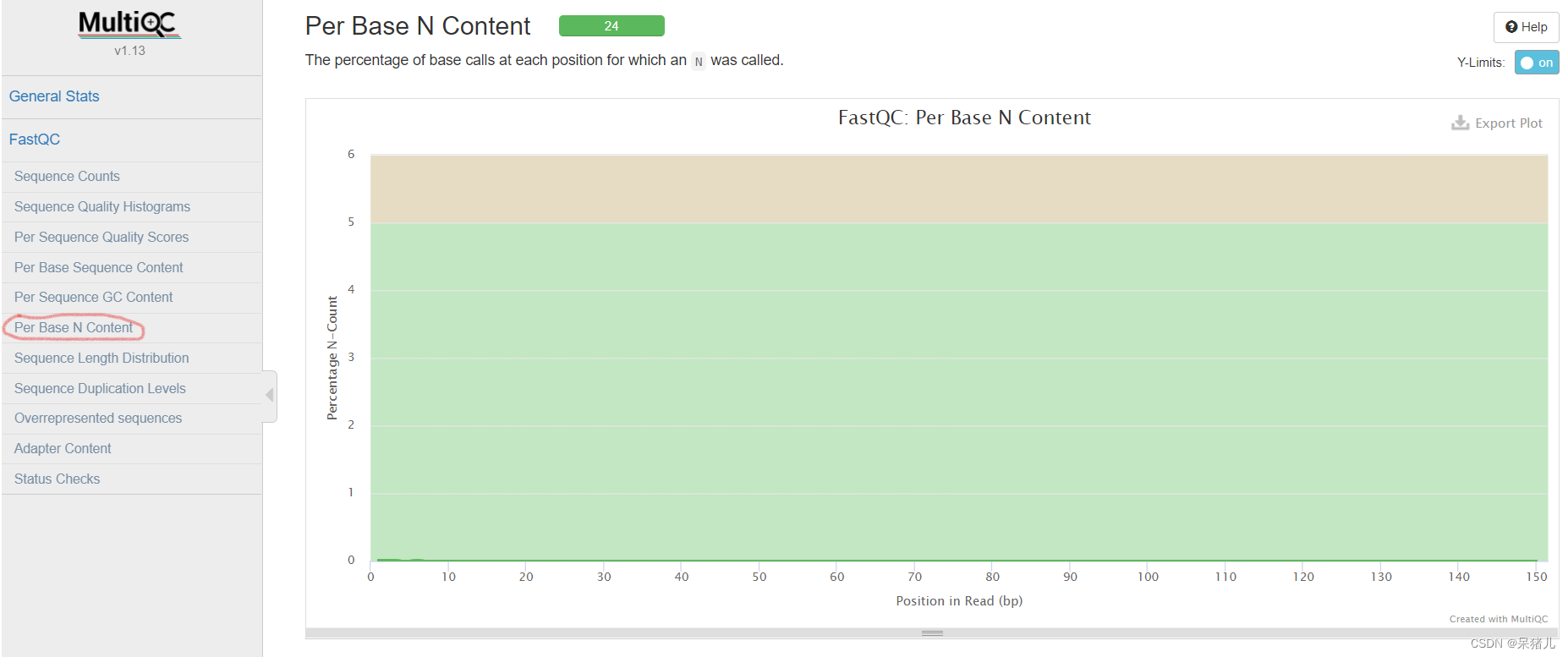 每条reads各位置N碱基含量比例 当不能辨别reads的某个位置到底是什么碱基时,就会产生一个N,所以N值越大就表明未知碱基越多 好的样本N值应该如图所示,N值很小,基本为0。
每条reads各位置N碱基含量比例 当不能辨别reads的某个位置到底是什么碱基时,就会产生一个N,所以N值越大就表明未知碱基越多 好的样本N值应该如图所示,N值很小,基本为0。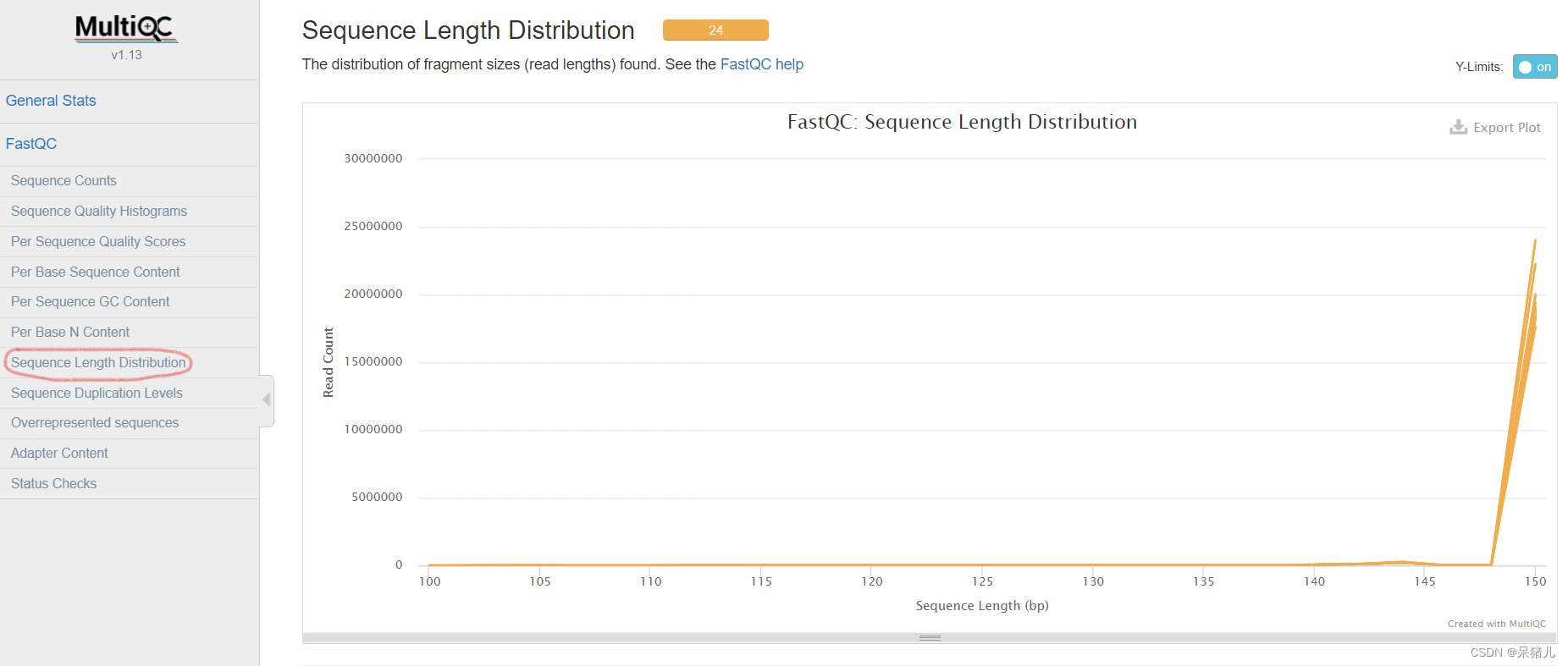 序列长度分布 这里主要显示测序长度和read counts的对应关系,我这里12个样本测序长度是150bp,对应的read counts基本都在15000000~20000000。
序列长度分布 这里主要显示测序长度和read counts的对应关系,我这里12个样本测序长度是150bp,对应的read counts基本都在15000000~20000000。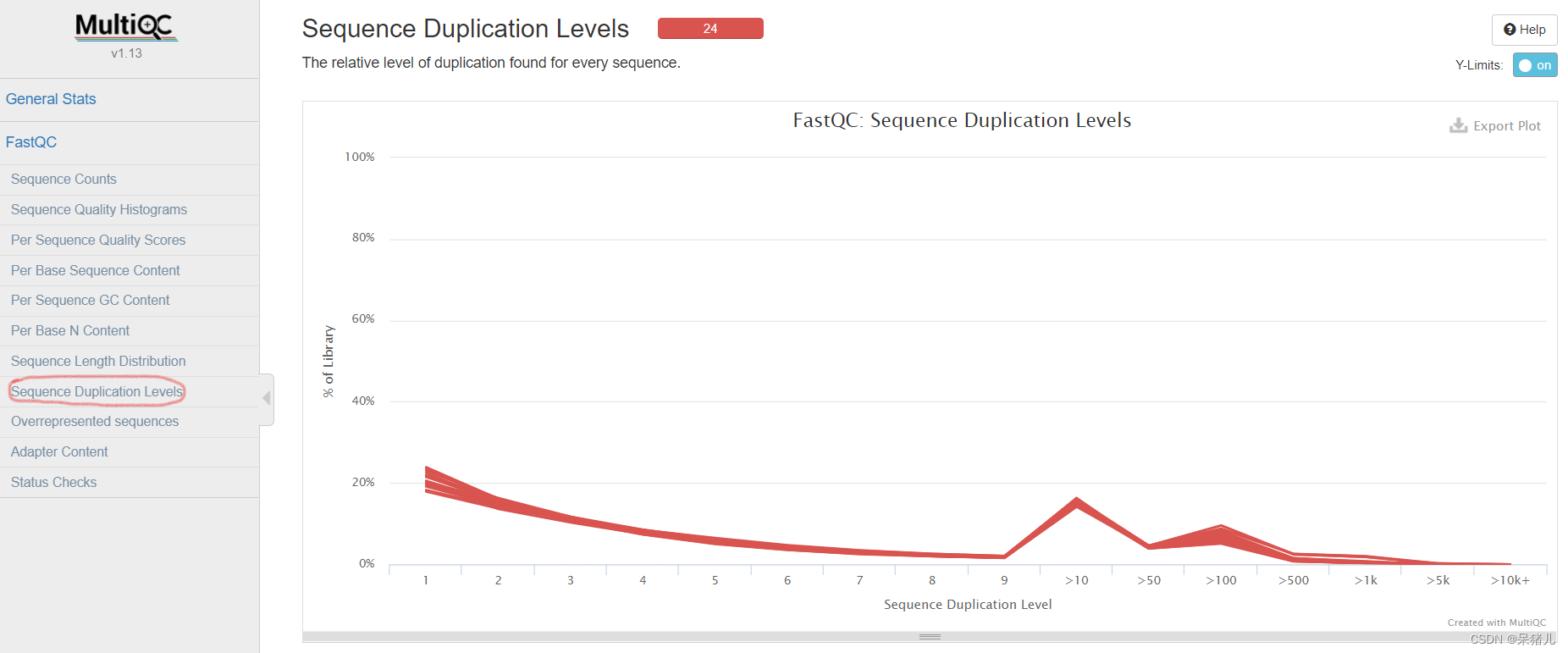 序列的相对重复水平 序列重复水平分布是统计序列完全一样的Reads的频率,通过该指标可以提示我们测序过程产生的偏差
序列的相对重复水平 序列重复水平分布是统计序列完全一样的Reads的频率,通过该指标可以提示我们测序过程产生的偏差
 我们可以看到质量过滤结果的详细信息 (1)代表的是质量分数阈值,前面代码设置的是25,这里自然就是25,表示质量小于25的都会被去除。 (2)可以忍受的前后接头重叠的碱基数,由于参数设定的是1,所以这里显示的是1bp。 (3)表示输出reads长度阈值,参数设定的是50,代表长度小于50的reads都会被去除。 (4)完成质量过滤所消耗的时间。 (5)表示接头重叠碱基数大于1的reads,我这里显示有36.4%的reads接头重叠碱基数大于1。 (6)最后一个表示质量小于25的占有0.3%,自然,这一部分会被去除(如果quality参数设的越大,这个值就会越大)。
我们可以看到质量过滤结果的详细信息 (1)代表的是质量分数阈值,前面代码设置的是25,这里自然就是25,表示质量小于25的都会被去除。 (2)可以忍受的前后接头重叠的碱基数,由于参数设定的是1,所以这里显示的是1bp。 (3)表示输出reads长度阈值,参数设定的是50,代表长度小于50的reads都会被去除。 (4)完成质量过滤所消耗的时间。 (5)表示接头重叠碱基数大于1的reads,我这里显示有36.4%的reads接头重叠碱基数大于1。 (6)最后一个表示质量小于25的占有0.3%,自然,这一部分会被去除(如果quality参数设的越大,这个值就会越大)。【本文地址】
今日新闻 |
点击排行 |
|
推荐新闻 |
图片新闻 |
|
专题文章 |