bam/sam文件格式介绍 |
您所在的位置:网站首页 › 第六列第十行可以表示为什么 › bam/sam文件格式介绍 |
bam/sam文件格式介绍
|
bam/sam文件格式介绍
SAM存储格式发明的目的:使得不同平台下机数据,经过不同比对软件后有一个统一的存储格式。
SAM(Sequence Alignment/Map format简写)格式文件,存储测序数据和参考基因组比对结果的文件,每行以table键分割,包含标头部分(header section)和比对部分(alignment section)见下图。
术语概念 模板(Template):一段DNA/RNA序列,它的一部分在测序仪上被测序,或被从原始序列中组装。(意思就是:我们通过测序仪测序的那段序列,或者通过组装原始序列得到的更长的序列,就是模板的一部分)。(从后文来看,对于Illumina双端测序来说,template指的就是插入片段) 片段(Segment):一段连续的序列或子序列(subsequence)(从上下文来看,segment既可以指一条完整的read,也可以指read的一部分); 读段(Read):一段来自测序仪的原始序列。read可以包含多个片段(一条read在比对过程中可能会被拆分成几段,对应到参考序列不同的位置上。read被拆分后形成的片段即为segment)。对于测序数据,reads根据测序顺序进行编号; 线性比对(Linear alignment):一条比对到参考序列上的read可能会有插入、缺失、skips和切除(clipping),但只要没有方向的改变(例如,read的一部分比对到了正义链上,另一部分比对到了反义链上),就是Linear alignment。一个线性比对结果可以代表一个SAM记录;(意思似乎是:一条SAM记录能且只能保存一个线性比对结果) 嵌合比对(Chimeric alignment):不是线性比对的比对。嵌合比对中包含了一套没有大范围重叠的线性比对(嵌合比对中的每一个片段都是线性比对。关于大范围重叠的说法是为了和多重比对区分)。一般地,嵌合比对中的一个线性比对被认为是“有代表性的比对”(representative alignment),而其他的线性比对被称为补充的(supplementary),用补充比对标志(supplementary alignment flag)加以区别(representative和supplementary成一对,对应嵌合比对)。嵌合比对的所有SAM记录有相同的QNAME,其flag值的0x40和0x80位都相同(见1.4节)(0x40位和0x80位分别表示模板中的第一个片段和最后一个片段,为什么会都相同呢?总要有一个是第一个片段,总要有一个是最后一个片段吧,它俩的0x40位和0x80位不应该相同啊?)。哪个线性比对被视为有代表性是任意选择的。(可见嵌合比对中,各个segments的独立性更强:都不在双链的同一条链了。另外,如果一条read的不同部分比对到了不同的染色体上,那肯定也是嵌合比对了,因为不同染色体之间讨论方向相同是没有意义的,肯定不可能是线性比对了。) read比对(read alignment):能代表一条read的比对结果的线性比对或嵌合比对; 多重比对(Multiple mapping):由于重复序列等情况的存在,一条read在参考基因组上的正确位置可能无法确定。在这种情况下,一条read可能会有多种比对结果,其中一种被视为主要的(primary),所有其他的比对结果的SAM记录的flag标志中都会有一个“次要(secondary)比对结果”的标志。所有这些SAM记录拥有相同的QNAME,flag标志的0x40位和0x80位有相同的值。一般被指定为“主要”的比对结果是最佳比对,如果都是最佳比对,则任意指定一条(primary和secondary成一对,对应多重比对)。(原文注释:嵌合比对主要由结构变异、基因融合、组装错误、RNA测序或实验过程中的一些原因造成,更经常出现在长reads中(长read有利于检测嵌合比对。这就是为什么三代测序是检测染色体结构变异的更有力工具)。嵌合比对中的线性比对之间没有大片段的重叠,每个线性比对有较高的mapping质量值,可以用于SNP/INDEL的检测;而多重比对主要是序列重复造成的,不经常出现在长reads中。如果一条read有多重比对的情况,所有的比对互相之间几乎完全完全重叠。除了一个最佳比对外,所有其他比对的质量值都< 3,且会被大多数SNP/INDEL检测软件忽略)。 以1为起始的坐标系(1-based coordinate system):序列的第一位是1的坐标系。在这种坐标系中,一个区域用闭区间表示。例如,第三位和第七位碱基之间的区域表示为[3,7]。SAM, VCF, GFF和Wiggle格式使用以1为起始的坐标系; 以0为起始的坐标系(0-based coordinate system):序列的第一位是0的坐标系。在这种坐标系中,一个区域用左闭右开区间表示。例如,第三位和第七位碱基之间的区域表示为[2,7)(原文如此。难道不应该是[3,8)么?不应该。以0为起始,第三位对应的索引号是2,第七位对应的索引号是6,所以索引号[2,7)对应了第三位-第七位碱基。当时脑子糊涂了,没搞清文中说的意思)。BAM, BCFv2, BED和PSL格式使用以0为起始的坐标系; Phred scale:如果一个概率值0 < p ≤ 1,这个p值的phred scale等于-10log(10)p,舍入为最近的整数。 一、sam文件header部分
这一部分就看看下表,不多介绍。
二、sam文件比对部分
该部分是SAM文件的核心部分,每一行代表一个序列的线性比对(linear alignment of a segment),每行包含前11个必需字段,和第12个字段后多个可选字段,使用TAB-separated分割,当某个字段信息缺省时,如果字段是字符串型以*替代,如果字段是整型以‘0’来替代,下表为11个必需字段含义的概述。
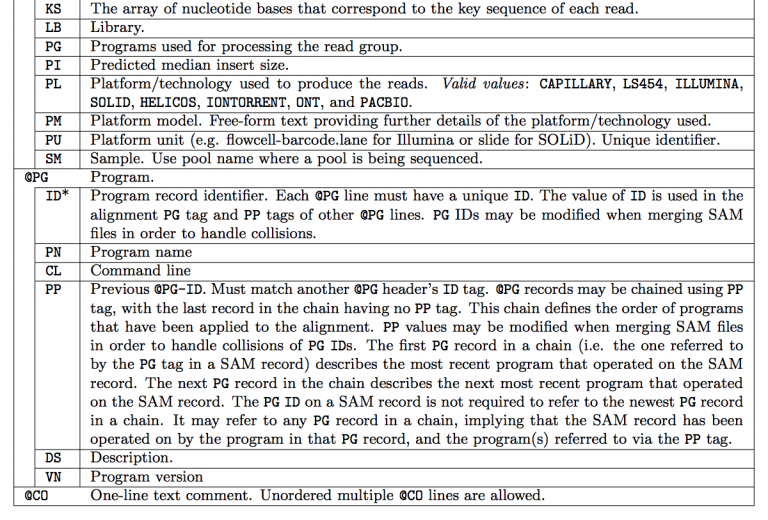
三、比对部分详述 第一列:QNAME 被比对序列的名称(query template name),如果QNAME唯一,则序列被认为来源于同一模板;‘*’表示该字段缺省;一般情况下,该字段为FASTQ文件的第一行信息;嵌合(Chimeric alignment)比对或者多次比对(Multiple mapping)的序列会导致一个QNAME在SAM中多次出现。 第二列:FLAG
第三列:RNAME Reference sequence NAME of the alignment,比对时参考序列的名称,一般是染色体号(如果物种为人,则为chr1~chr22,chrX,chrY,chrM)。RNAME(如果不是“*”)必须在header section部分@SQ中SN标签后出现。如果没有比对上参考基因组,用"*"来表示。如果RNAME值是"*",则后面POS和CIGAR也将没有值。 第四列:POS 该read比对到参考基因组的位置坐标,最小为1(1-based leftmost)。该read如果没有比对上参考序列,则RNAME和CIGAR也无值。 第五列:MAPQ 对应参考序列的质量(MAPing Quality),比对的质量分数,越高说明该read比对到参考基因组上的位置越准确。其值等于-10 lg Probility (错配概率),得出值后四舍五入的整数就是MAPQ值。如果该值是255,则说明对应质量无效。例如,MAPQ为20,即Q20,错误率为0.01,20 = -10log10(0.01) = -10*(-2)。 第六列:CIGAR
Compact Idiosyncratic Gapped Alignment Representation的简写,描述read与参考序列的比对具体情况信息。CIGAR中的数字代表碱基的个数,字符的含义见下表:
第七列:RNEXT 双端测序中另外一条read比对的参考序列的名称,单端测序此处为0,RNEXT(如果不是*或者=,*是完全没有比对上,=是完全比对)必须在header section部分@SQ中SN标签后出现。第3和第7列,可以用来判断某条read是否比对成功到了参考序列上,read1和read2是否比对到同一条参考染色体上。 第八列:PNEXT 双端测序中,是指另外一条read比对到参考基因组的位置坐标,最小为1(1-based leftmost)。 第九列:TLEN
文库长度,insert DNA size。
第十列:SEQ read 碱基序列,FASTQ的第二行。 第十一列:QUAL FASTQ的第四行。 第十二列之后,Optional fields 可选的自定义区域(Optional fields),可能有多列,多列间使用\t隔开,并不是每行都存在这些列。 References: https://learn.gencore.bio.nyu.edu/ngs-file-formats/sambam-format/ https://ming-lian.github.io/2019/02/07/Advanced-knowledge-of-SAM/ https://blog.csdn.net/qq_21478261/article/details/106005482 https://www.cnblogs.com/timeisbiggestboss/p/8856888.html |
 BAM(Binary Alignment/Map format简写)格式文件,SAM的二进制格式文件,通过BGZF library参考库压缩而成。
BAM(Binary Alignment/Map format简写)格式文件,SAM的二进制格式文件,通过BGZF library参考库压缩而成。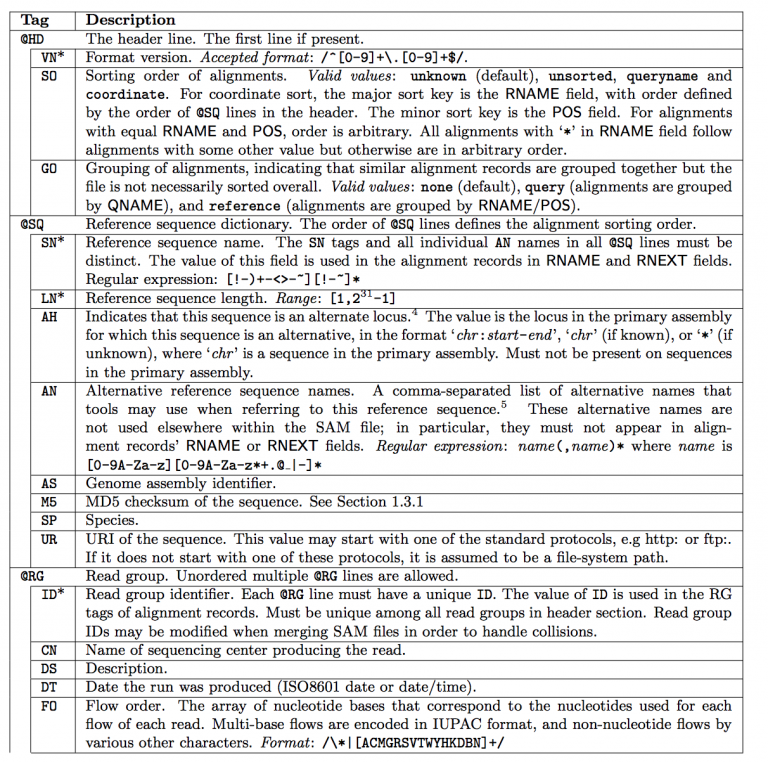

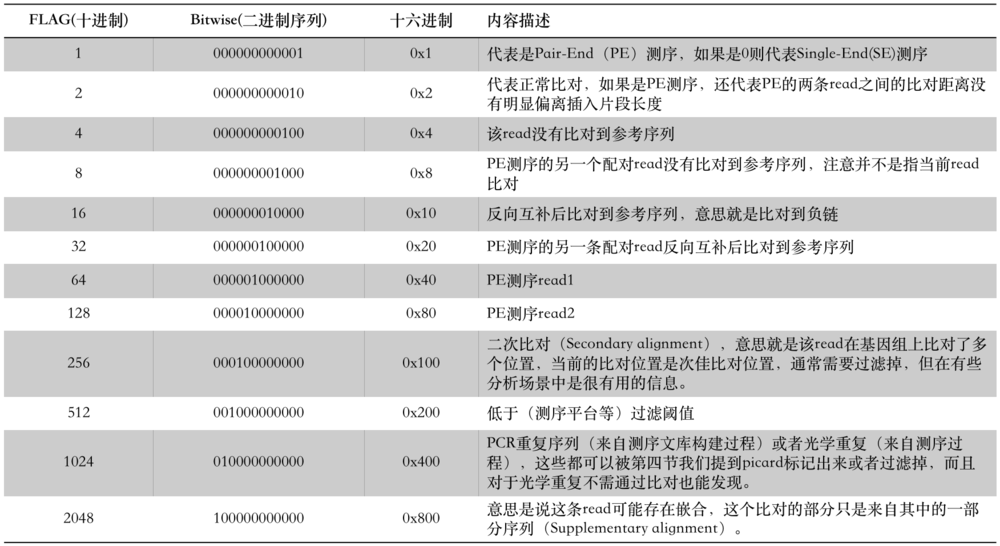 实际情况,一条read的比对往往包含以上情况的一种或一种以上,所以对应的FLAG值也是几种情况FLAG值的加和。同样,我们也可以根据该read的FLAG值,反推出其对应的比对情况。这里有以下几个网站可利用进行查询:
http://https://broadinstitute.github.io/picard/explain-flags.html
https://www.samformat.info/sam-format-flag
FLAG值常常会用到reads alignment过滤中,所以对它的理解是尤为重要的。
其中涉及到reads重复比对的几点可能稍微难以理解:
实际情况,一条read的比对往往包含以上情况的一种或一种以上,所以对应的FLAG值也是几种情况FLAG值的加和。同样,我们也可以根据该read的FLAG值,反推出其对应的比对情况。这里有以下几个网站可利用进行查询:
http://https://broadinstitute.github.io/picard/explain-flags.html
https://www.samformat.info/sam-format-flag
FLAG值常常会用到reads alignment过滤中,所以对它的理解是尤为重要的。
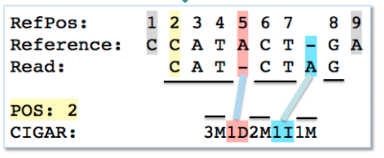
其中涉及到reads重复比对的几点可能稍微难以理解: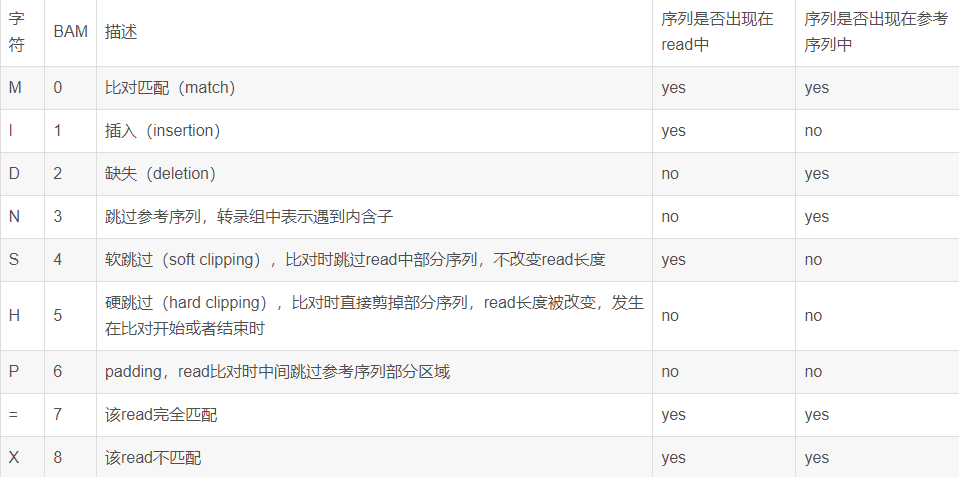 举例:3M1D2M1I1M:3个碱基匹配(M)(3M)、接下来1个碱基缺失(D)、接下来2个匹配(2M)、接下来1个碱基插入(1I)、接下来1个碱基匹配(1M),如下图:
举例:3M1D2M1I1M:3个碱基匹配(M)(3M)、接下来1个碱基缺失(D)、接下来2个匹配(2M)、接下来1个碱基插入(1I)、接下来1个碱基匹配(1M),如下图:

【本文地址】
今日新闻 |
点击排行 |
|
推荐新闻 |
图片新闻 |
|
专题文章 |