靶向热休克蛋白90的双功能小分子抑制剂的抗肿瘤研究进展 |
您所在的位置:网站首页 › 抑制蛋白的作用有哪些 › 靶向热休克蛋白90的双功能小分子抑制剂的抗肿瘤研究进展 |
靶向热休克蛋白90的双功能小分子抑制剂的抗肿瘤研究进展
|
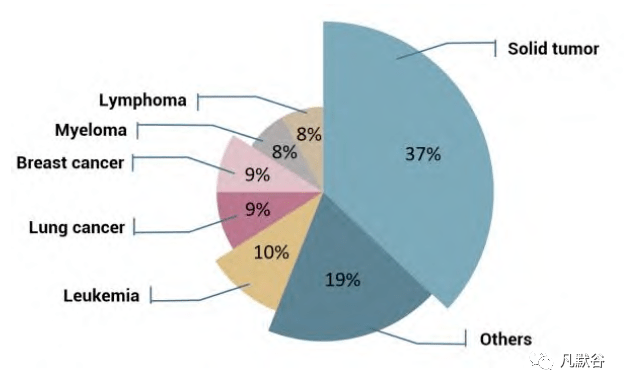
Hsp90的过度表达通常是为了减轻细胞损伤以应对各种细胞压力,导致客户蛋白的错误折叠和错误积累,影响信号转导网络,最终导致生理功能异常、细胞内蛋白质动态平衡失衡,以及复杂的疾病,如癌症、神经退行性疾病和免疫系统疾病[4-6]。因此,靶向Hsp90和Hsp90-客户复合体一直被认为是治疗各种疾病的潜在方法[7-9],特别是癌症[10-12]。自从1999年17-烯丙氨基-17去甲氧基格尔达霉素(17-AAG)作为一类Hsp90抑制剂进入临床试验以来,已经开展了100多项Hsp90抑制剂的肿瘤学试验,涉及各种类型的癌症,最常见的适应症是广谱肿瘤(图2)[12-15]。因此,开发Hsp90抑制策略和发现小分子Hsp90抑制剂一直是医药和化学领域关注的主要领域,许多研究小组和制药公司在研究基于Hsp90的新药方面做出了实质性的尝试[16-19]。
Figure 2 Proportion of clinical trials of cancer types involving Hsp90 inhibitors (from ClinicalTrials.gov) 然而,进入临床试验的大多数Hsp90抑制剂由于疗效低、毒性或耐药性而被宣布临床失败。尽管进入临床试验的Hsp90抑制剂的数量在过去10年中有所减少,但Hsp90抑制剂与其他抗肿瘤药物或癌蛋白抑制剂的药物组合比例明显增加[20-22]。临床试验表明,Hsp90抑制剂与其他抑制剂的组合具有协同作用和抗耐受的特性。鉴于Hsp90抑制和联合治疗的重要意义,开发Hsp90双重抑制剂引起了科学领域的关注,2020年相关论文总数呈现爆发式增长。 本文综述了Hsp90的结构域结构和生物学功能,并对Hsp90双抑制剂的设计、发现和构效关系进行了讨论,旨在从药物化学的角度为临床抗肿瘤药物的研究开发提供启示,并为发现新的Hsp90双重抑制剂奠定基础。 1 Hsp90抑制剂的结构域结构、生物学功能及双靶点设计 1.1 Hsp90的结构域结构及其相应的抑制剂
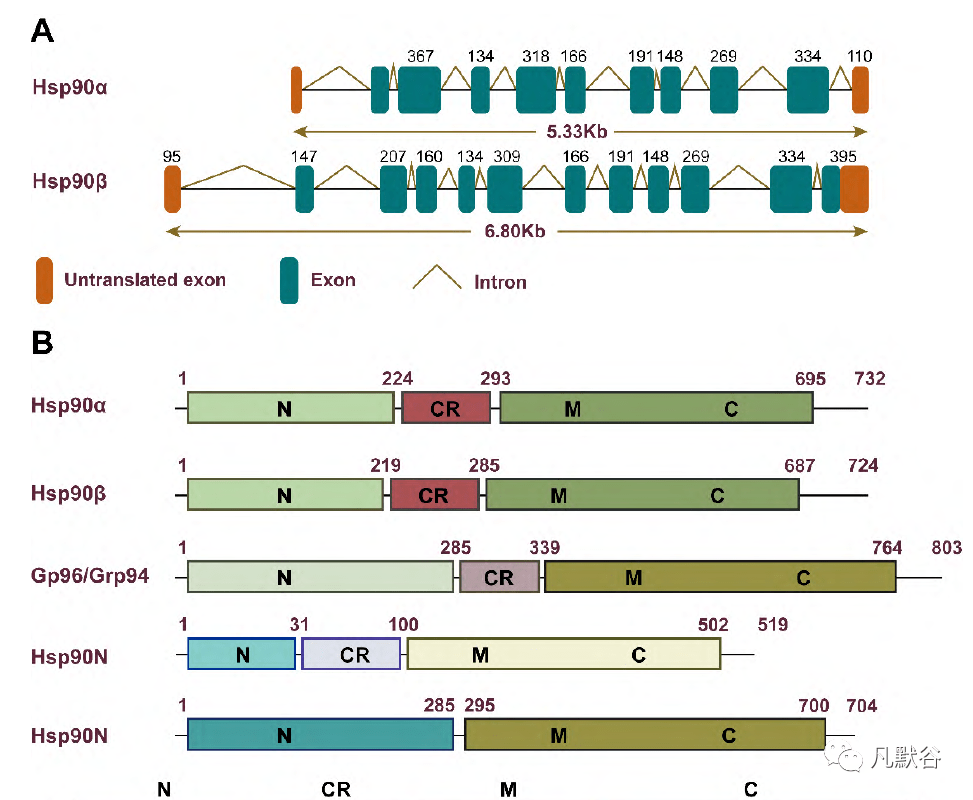
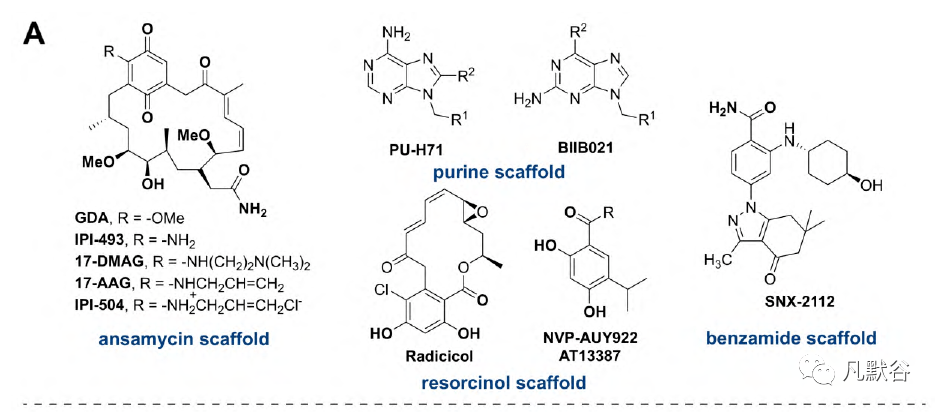
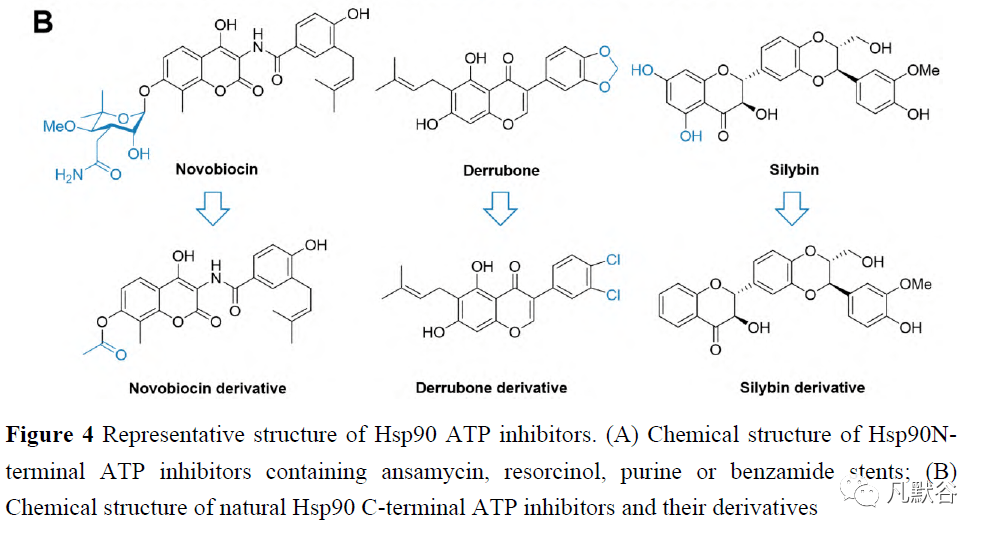
位于N-末端结构域的ATP位点已经引起了人们的极大兴趣,并被大量研究用于开发Hsp90抑制剂,其中一些已经进入临床试验。经典的结构如阿萨霉素(ansamycin)、嘌呤(purine)、间苯二酚(resorcinol)和苯甲酰胺(benzamide),是必不可少的Hsp90N竞争抑制剂(图4A)[5, 29-31]。C端域作为ATP结合口袋,被天然产物新生霉素(novobiocin,第一个Hsp90C抑制剂)和其他天然产物,如鱼藤素(deguelin)、地龙骨(derrubone)和水飞蓟宾(silybin)靶标。研究表明,经过结构修饰这些天然产物的衍生物对Hsp90C抑制有效(图4B)[32-36]。
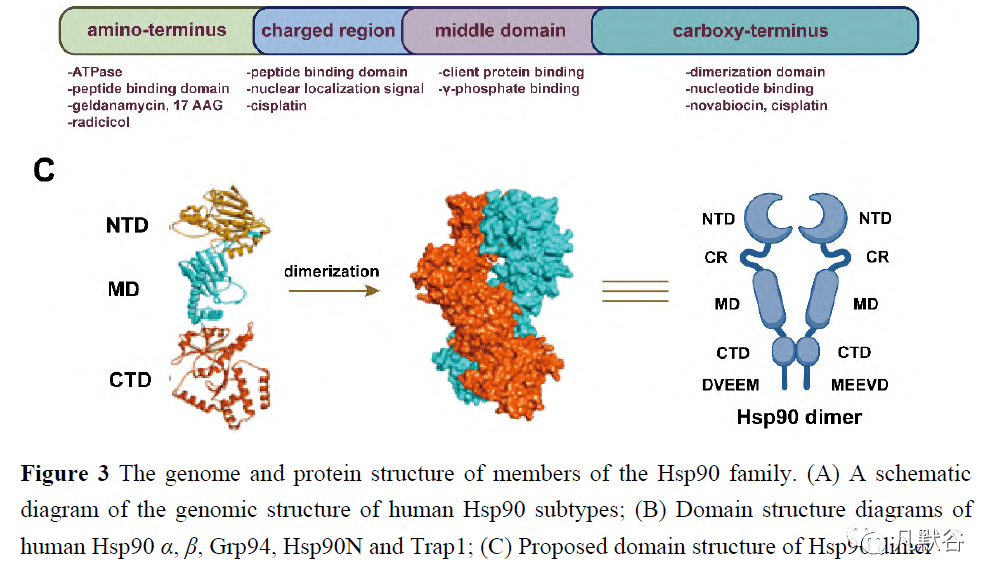
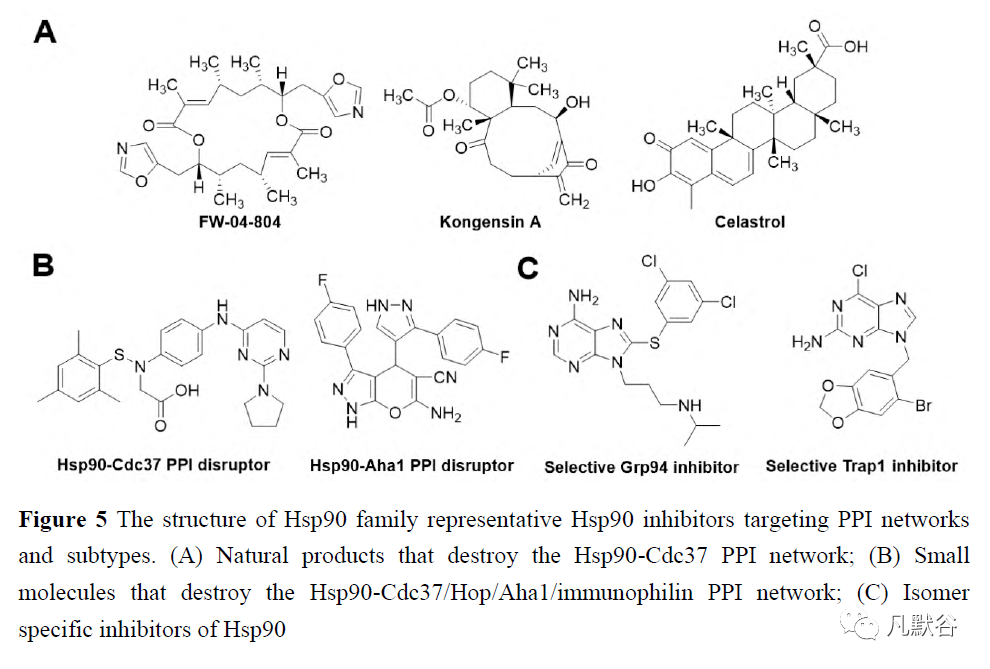
一些Hsp90抑制剂的另一种抑制机制是直接破坏Hsp90与辅助伴侣之间的相互作用,而Hsp90充当分子伴侣。天然产物如雷公藤红素(celastrol)、睡茄素A(kongensin A)、萝卜硫素(sulforaphane)、FW-04-804[37, 38],以及一些小分子,都是Hsp90-Cdc37蛋白-蛋白相互作用(protein-protein interaction, PPI)的抑制剂,但它们没有共同的骨架(图5A、B)[39-41]。嘧啶并[5, 4-e][1, 2, 4]-三嗪-5, 7-二酮结构核心对于破坏Hsp90-Hop-Hsp70复合物至关重要(图5B)[42]。单个分子被确定为Hsp90-亲和蛋白及Hsp90-Aha1PPI网络的干扰物(图5B)[43, 44]。 Hsp90的亚型特异性抑制和随后相关的药理学和细胞通路影响各种客户蛋白的功能。Hsp90是一种大的构象动态蛋白,根据构型在哺乳动物细胞中可分为4类:诱导型Hsp90α和组成型活性蛋白胞质溶胶中的Hsp90β、内质网中的Grp94[45]、以及与线粒体相关的Trap1[46]。与Hsp90α/β序列相比,Grp94的N端多出5个残基QEDGQ,C端多出KDEL,这对内质网定位至关重要,而Trap1的N端多出一个线粒体信号序列。Hsp90α/β中N端结构域和C端结构域之间的带电接头负责核定位并调节Grp94中的钙活性。异构体特异性抑制剂的开发是根据结构和构象变化而不是类似的核苷酸结合域(图5C)[47-49]。
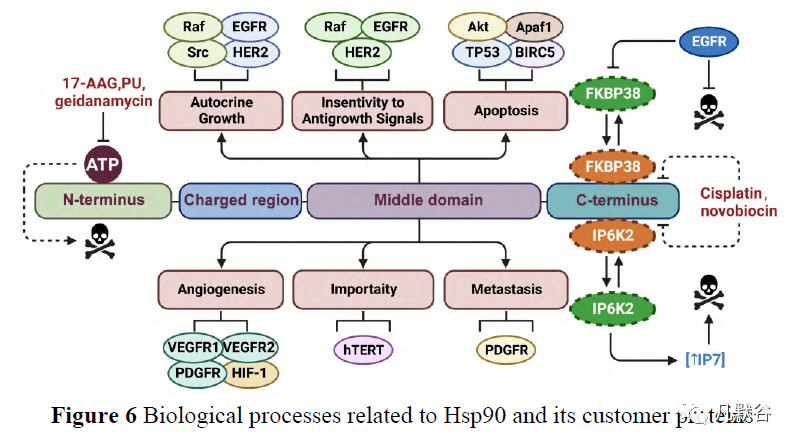
1.2 Hsp90的生物学功能 在正常细胞中,低细胞质Hsp90β表达足以在细胞周期、增殖、信号转导和转录过程中调节细胞稳态。然而,在重金属暴露、高温和氧化应激等不同类型的压力下,Hsp90α会上调并促进许多客户蛋白的成熟,包括致癌激酶[4]。Hsp90α和Hsp90β调节Tau、人表皮生长因子受体2(human epidermal growth factor receptor 2, HER2)、Akt、表皮生长因子受体(epidermal growth factor receptor, EGFR)和细胞周期蛋白依赖性激酶4或6(cyclin-dependent kinases 4 and 6, CDK4/6)。Grp94主要调节整合素、HER2、LRP6、Toll样受体和Wnt/β-catenin信号通路[50, 51]。Wnt/β-连环蛋白信号通路对肿瘤形成和免疫调节至关重要,但在Grp94敲除肿瘤细胞系中被阻断[52]。Trap1主要调节细胞周期停滞并降低与细胞增殖和凋亡相关的活性氧(reactive oxygen species, ROS)水平[53]。一方面,Hsp90的过表达引起客户蛋白的过度积累,加速细胞增殖、迁移和侵袭,促进血管生成和细胞周期;另一方面,Hsp90的功能依赖于辅伴侣辅助的ATP水解和客户蛋白成熟。Co-chaperones的主要功能是调节Hsp90ATP的活性,从而进一步改变Hsp90的构象,并聚集特异性组装在Hsp90上的未成熟客户蛋白[54],与Hsp90及其客户蛋白相关的生物过程如图6所示。
1.3 与Hsp90抑制剂相关的药物组合 作为有希望的癌症治疗选择,一些Hsp90抑制剂正在进行临床试验[7]。然而,作为单一疗法的Hsp90抑制剂在临床阶段存在一些不理想的结果,包括疗效不佳、耐药性以及无法耐受的不良反应,如肝毒性、眼毒性和心脏毒性[26]。由于Hsp90抑制剂和其他化疗药物具有协同作用,将两者组合已成为研发抗肿瘤药物的新策略。 1.3.1 Hsp90抑制剂与PI3K-Akt-mTOR信号通路抑制剂的药物组合 在Hsp90和其他肿瘤相关蛋白的所有多靶向案例中,靶向Hsp90及其下游途径是克服耐药潜力最大的策略。PI3K-Akt-mTOR是Hsp90下游信号之一,在常见的癌症类型中普遍过度激活,因此研究者试图同时抑制该途径和Hsp90。NVP-BEZ235(PI3Kt-mTOR抑制剂)与多种Hsp90抑制剂联合使用时,对受试细胞或相应的动物模型具有协同抗肿瘤作用。17-DMAG(Hsp90抑制剂)和NVP-BEZ235联合应用对顺铂耐药的膀胱癌显示出协同的抗肿瘤作用,诱导细胞周期停滞于G1期,并诱导半胱氨酸酶依赖的细胞凋亡[55]。 NVP-AUY922(Hsp90抑制剂)和NVP-BEZ235在诱导肝内胆管癌细胞(cholangiocarcinoma, CCA)细胞死亡和导致CCA动物模型肿瘤消退方面具有协同作用,这种协同作用与ROS诱导有关[56]。17-AAG(Hsp90抑制剂)和NVP-BEZ235的组合在抑制黑色素瘤细胞生长、诱导细胞凋亡以及同时靶向MAPK和PI3K-Akt-mTOR通路方面具有协同作用[20]。此外,17-AAG和Torin2(mTOR抑制剂)在体内和体外协同抑制间变性甲状腺癌(anaplastic thyroid carcinoma, ATC)的生长[57]。 1.3.2 Hsp90抑制剂与HDAC抑制剂的药物组合 由于组蛋白脱乙酰酶(histone deacetylase, HDAC)抑制剂可阻断ATP结合,使Hsp90的伴侣功能失效,并使对Hsp90抑制剂产生耐药性的耐药细胞重新敏感,目前已探索了PAN HDAC和选择性HDAC6抑制剂与Hsp90抑制剂之间的协同作用[58, 59]。贝力诺他是一种HDAC抑制剂,常与其他药物联合治疗多种肿瘤。在三阴性乳腺癌MDA-MB-231细胞中,17-AAG和贝力诺斯特具有协同作用。这些效应在治疗后的增殖、凋亡和细胞周期停滞过程中被检测到[58, 60]。另外,AUY922和贝利司他在ATC细胞死亡中观察到协同作用[58];HDAC抑制剂伏立诺他(SAHA)和曲古菌素A(TSA)与贝力诺斯特具有协同作用并诱导ATC细胞的细胞毒作用,这种作用伴随着PI3K-Akt-mTOR信号和生存素的抑制,以及DNA损伤相关蛋白的过度表达[59]。 1.3.3 Hsp90抑制剂与其他靶向抑制剂的药物组合 1.4 Hsp90双重靶向抑制剂的设计
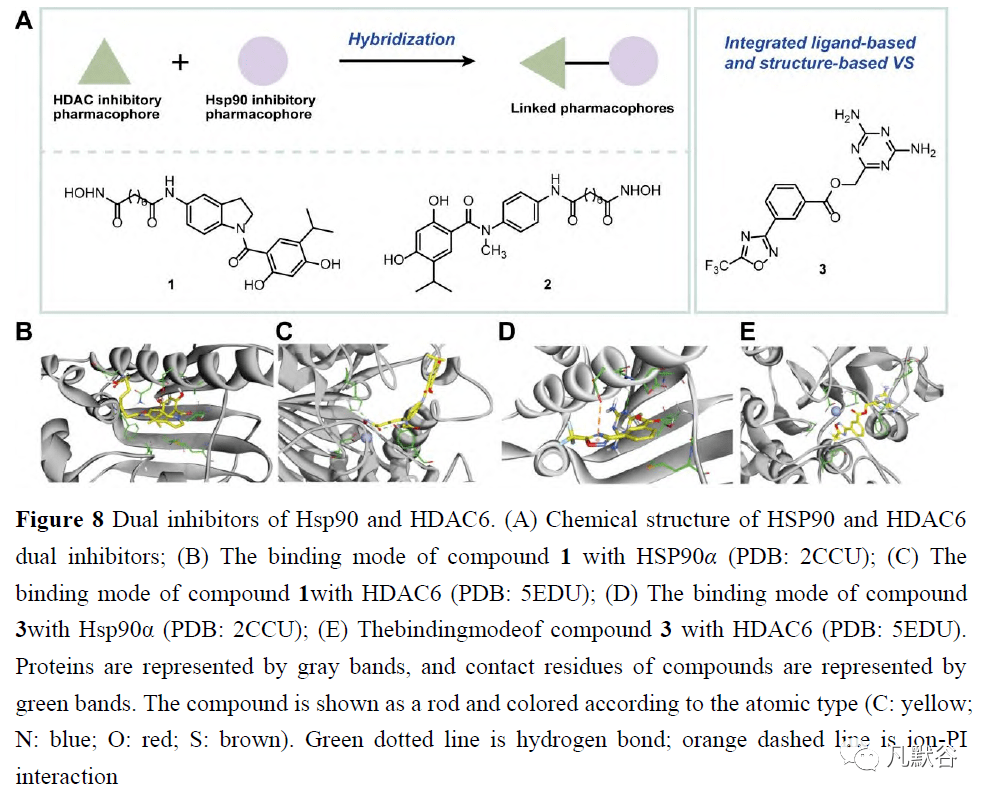
文章内容由凡默谷小编查阅文献选取,排版与编辑为原创。如转载,请尊重劳动成果, 注明【来源:凡默谷公众号】。 2 Hsp90抑制剂和其他癌蛋白的双重抑制物 2.1 Hsp90和HDAC6的双重抑制剂 HDAC在表观遗传变异和基因表达改变中起重要作用,与许多癌基因和肿瘤抑制因子有关,因此被认为是潜在的治疗靶点[71, 72]。鉴于HDAC抑制剂影响Hsp90的伴侣功能,在Hsp90抑制剂和HDAC抑制剂的组合中观察到协同效应[73]。从2018年开始,刘景平团队开始致力于Hsp90/HDAC双重抑制剂的开发。他们设计了一种由先前报道的具有非平面吲哚核心的HDAC抑制剂和4-异丙基间苯二酚片段组成的混合骨架,该片段作为与Hsp90蛋白质的ATP结合位点相关的关键结合剂,也是第二代Hsp90抑制剂的基本结构[74]。一系列具有不同烷基长度的1-芳基吲哚-异羟肟酸(n=2~8)或吲哚环上C-5位的SAHA衍生的N-苄基接头具有细胞毒性(图8)。
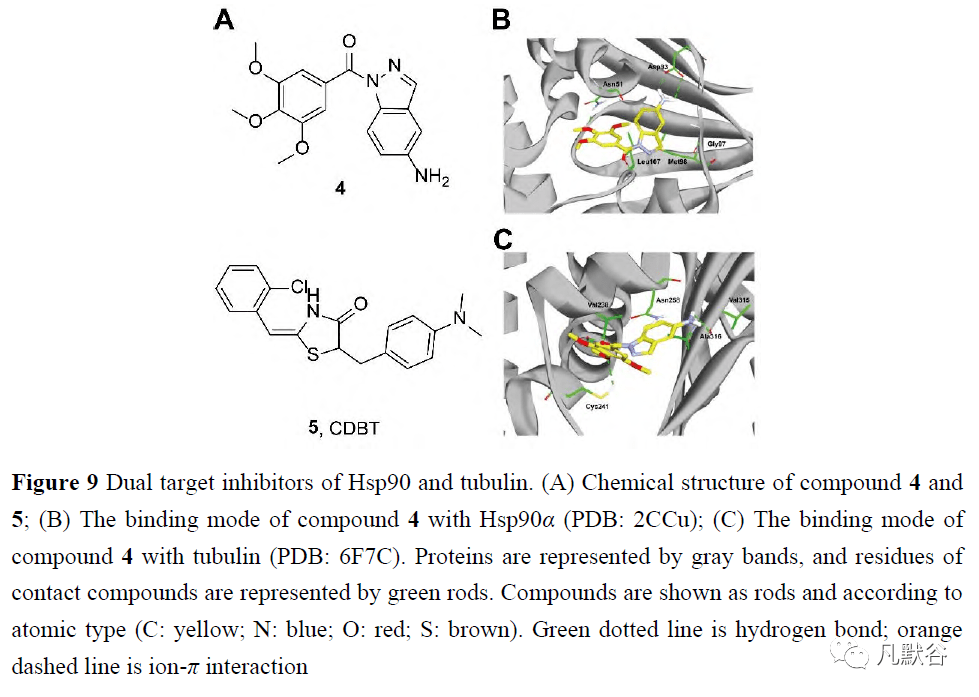
化合物1对Hsp90和HDAC表现出最佳的双重抑制作用(图8A)。在吲哚核心上与SAHA相同的连接基和锌结合基团的组合下,化合物1显示出比FDA批准的HDAC抑制剂SAHA高1.6倍的HeLa HDAC抑制,以及比口服小分子Hsp90抑制剂BIIB021更高的Hsp90抑制。与此同时,多种Hsp90客户蛋白和HDAC相关蛋白,如Akt、Hsp70和STAT3,在一定浓度和时间内下调结果表明,化合物1通过对Hsp90和HDAC的双重抑制而对癌细胞产生细胞毒作用。对HDAC异构体酶抑制的进一步研究表明,HDAC6异构体对1-芳基吲哚异羟肟酸最敏感,尤其是化合物1,甚至比参比化合物TSA更敏感。 刘景平团队在2019年开发了一系列Hsp90和HDAC的双重抑制剂,基于2006年被FDA批准为HDAC抑制剂的SAHA的结构和间苯二酚部分,后者被确定为适合设计Hsp90抑制剂的片段[75]。以相应的丁氧基氨基甲酸苯胺为底物合成了各种羟基甲酸酯类化合物2。一系列新的异羟肟酸2是组蛋白的双重抑制剂,尤其是那些侧链含有7个碳烷基的化合物,其中烷基化合物表现出比未烷基化合物更好的抑制活性。化合物2对HDAC6的抑制作用最强,IC50值在纳摩尔范围内,对HDAC6异构体的选择性高于其他化合物。此外,它们还能以剂量依赖的方式减少干扰素-γ处理的肺H1975细胞中程序性死亡配体1的表达。 2020年,Pinzi等[76]利用集成计算方法设计了新型HDAC6和Hsp90双重抑制剂。由于Hsp90和HDAC6与它们自己的配体在结构和结合部位结构上几乎没有同源性,研究人员结合了基于硅配体和基于结构的VS方法来预测与这两个靶标结合的必要片段,并选择了10个对接分数可接受的候选片段,合成了所有的预测分子,并对其生物活性进行了评价。体外研究表明,它们通过调节多个靶点抑制MCF-7细胞的增殖。一些候选药物已被证明在同时抑制HDAC6和Hsp90方面有效。最有效的分子化合物3,能靶向于具有多个结合位点的HDAC6:它与5-(三氟甲基)-1, 2, 4-恶二唑部分配位锌离子,而具有π−π堆积作用的Phe620和Phe680残基由苯环、His500、Pro501和Leu749与2, 4-二氨基三嗪片段提供(图8D)。虽然化合物3不能有效地抑制Hsp90,但苯环部分符合Hsp90的Leu103、Leu107和Phe138残基,而5-(三氟甲基)-1, 2, 4-恶二唑环向由残基Phe22、Gly108、Trp162和Phe170排列的子袋延伸,从而进一步优化了结构(图8E)。 2.2 Hsp90与微管蛋白的双重抑制剂 Hsp90和微管是癌细胞增殖和存活的关键蛋白质,因此被认为是抗肿瘤的靶点,它们的生物学功能可能是协同联系的[77]。Hsp90保护微管蛋白免受热变性,并保持其有利于微管聚合的状态。Hsp90和微管蛋白抑制剂在体外和体内显示出协同抗肿瘤作用[78]。Hsp90和微管蛋白的双重抑制剂引起了人们的极大兴趣(图9)。 2009年,Knox等[79]集中研究了3′, 4′, 5′-三甲氧基苯基的药效团,它们来自3-芳基硫代吲哚(一种微管蛋白结合化合物)和PU3(一种与Hsp90结合的嘌呤类似物),PU3被认为是一个单一的支架,可以用于VS和鉴定与Hsp90或微管蛋白具有强大的双重结合亲和力的分子。一个基于配体和基于结构的VS工作流由对接或评分协议和基于受体的药效团组成,被设计用于检索“一流”分子1-[(3, 4, 5-三甲氧基苯基)-羰基]-1H-吲唑-5-胺(4, MDG892),它从约16万个化合物的商业光谱数据库中击中了这两个目标(图9A)。基于MCF-7细胞的Western blot分析表明,化合物4的200μmol·L -1 可通过直接结合Hsp90ATP结合部位而不是秋水仙素部位,从而诱导Hsp90客户蛋白雌激素受体R的降解。此外,对4在Hsp90中的结合模式的分析表明,4与Asp93形成了氢键,与Lys112形成了两个氢键,但没有观察到与Thr184的直接结合(图9B)。4与微管蛋白的结合模式表明,4与Ser178(3Å)形成了氢键,但与Thr179没有普遍的相互作用(图9C),为合成作为Hsp90/微管蛋白双重抑制剂的替代骨架提供了方向。
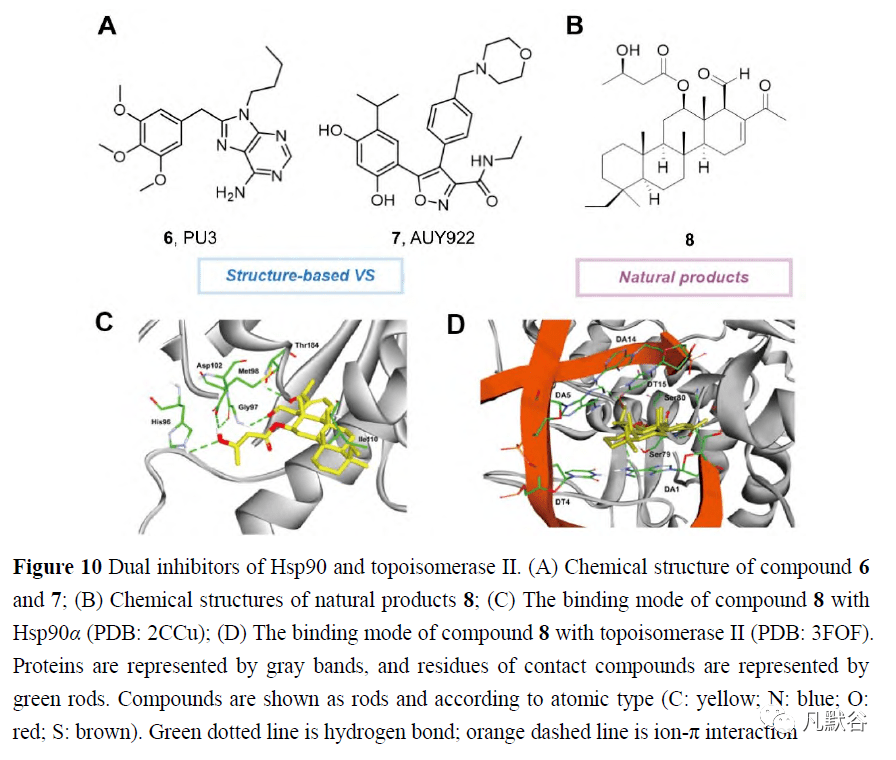
Zhang等[80]证明了CDBT在P-糖蛋白(P-gp)诱导的耐药NSCLC H460 TaxR 细胞中具有很强的抗肿瘤活性,并探讨了其作用机制。CDBT不是P-gp底物,这使其能够逃避P-gp介导的外排。作为Hsp90/微管蛋白的双重靶向剂,CDBT导致Hsp90的客户蛋白降解和微管解聚,导致细胞周期停滞在G2/M期,以及在H460 TaxR 细胞中由胱天蛋白酶8、9和3和PAPR裂解介导的凋亡。此外,CDBT单一疗法在人类NSCLC H460 TaxR 异种移植模型中产生60.4%的肿瘤生长抑制率。该值与在相同剂量水平和方案下其在亲代H460异种移植物模型中的功效(62.4%肿瘤生长抑制)相当。该研究提供了强有力的证据,证明了合成化合物可以发展成为非小细胞肺癌细胞的治疗剂,即使对具有耐药性的细胞也有抑制作用。 2.3 Hsp90和拓扑异构酶II的双重抑制剂 Topo II和Hsp90在增殖的癌细胞中同时过表达。此外,Topo II与Hsp90在结构上具有相似性,两个靶标包含属于同一家族的保守ATPase结构域,即GHKL(促旋酶、Hsp90、组氨酸激酶和MutL)结构域[81]。因此,Topo II和Hsp90的双重抑制是一个有希望的方向(图10)。
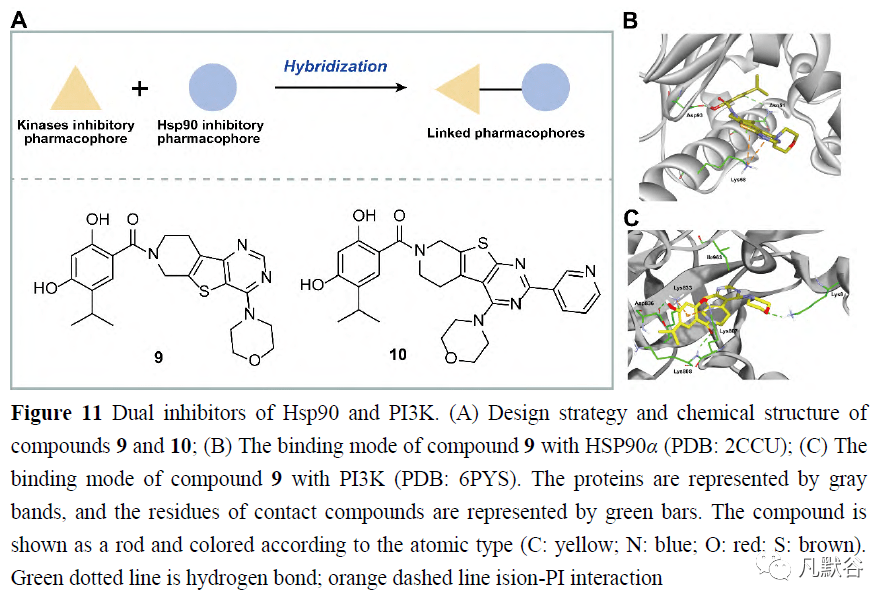
由于Hsp90/Topo II的双重抑制在癌症治疗中的重要性,以及Hsp90和Topo II上相似的ATPase结构域,Jun等[82]在2016年试图在比较两种靶蛋白的结构相似性的基础上,开发靶向Topo II和Hsp90的ATPase结构域的新型抑制剂。尽管两个ATPase结构域之间的序列同一性较低(15.8%),但它们的ATPase结合位点具有相似的环境,这有助于通过分子对接研究在已报道的仅具有Topo II或Hsp90抑制作用的分子中发现双重抑制剂。包括化合物6和7在内的4个候选物被选为最有效的双重抑制剂,并进一步用作药效团模型的模板(图10A),这可用于Hsp90和Topo II双重抑制剂的开发。 虽然还没有合成分子作为Hsp90和Topo II的双重抑制剂的报道,但是天然产物已有作为这两种蛋白的双重抑制剂。2016年,Lai等[83]从海绵Carteriospongia sp. 中分离得到了斯卡拉烷二萜类化合物12β-(3′β-羟基丁酰氧基)-20,24-二甲基-24-氧代-斯卡拉烷-16-en-25-al(8),并进一步证明了其在0.0625μg·mL -1 (125nmol·L -1 )浓度下,触发白血病Molt4细胞线粒体膜电位破坏和凋亡并抑制拓扑异构酶IIα表达的有效作用(图10B)。此外,12β-(3′β-羟基丁酰氧基)-20,24-二甲基-24-氧代-斯卡拉烷-16-en-25-al比标准的Hsp90抑制剂17-AAG对Hsp90蛋白的N-末端ATP结合口袋表现出更强的结合亲和力(图10C)。确定了其Hsp90抑制活性,某些Hsp90客户蛋白的表达,如Akt、p70S6k、NF-κB、Raf-1、p-GSK3β、XIAP、MDM2、Rb2、CDK4、细胞周期蛋白D3,HIF1和HSF1的表达被抑制,而Hsp70、乙酰化微管蛋白和活化的半胱天冬酶3的表达在治疗后被诱导。 2.4 Hsp90和PI3K的双重抑制剂 PI3K-Akt-mTOR是Hsp90下游信号之一,在常见的癌症类型中普遍过度激活,因此对PI3K-Akt-mTOR途径和Hsp90蛋白的双重抑制已成为Hsp90抑制剂药物组合研究的主要领域之一[20, 56, 84]。然而,用单分子对这种靶标的双重抑制剂很少被研究(图11)。 2019年,本团队[85]致力于发现Hsp90及其致癌客户蛋白的多靶点小分子抑制物,并报道了使用2, 4-二羟基-5-异丙基苯甲酸酯片段抑制Hsp90和硫代并[2, 3-d]嘧啶骨架抑制酶获得了首个Hsp90/磷脂酰肌醇3K双抑制剂。设计并合成了一系列2, 4-二羟基-5-异丙基苯甲酸酯衍生物,作为一种新型的Hsp90/PI3K双重抑制物在B16黑色素瘤细胞中的应用(图11A)。构效关系分析表明,4-烷基化取代比4-芳香取代对Hsp90的抑制作用更有利。化合物9对HSP90和PI3K的抑制作用最强,IC50值在纳摩尔水平,可抑制黑色素瘤细胞的迁移、侵袭和增殖,诱导细胞凋亡和细胞周期停滞,调节PI3K-Akt信号和Hsp90客户蛋白及其下游效应分子。分子对接研究表明,化合物9通过2,4-二羟基苯甲酸酯与Asp93形成稳定的氢键,从而与Hsp90进行结合(图11B);通过硫代并[2, 3-d]嘧啶与Lys58的阳离子相互作用,与PI3K通过3个稳定的氢键与Lys802、Ala805和Val882结合,并通过4-二羟基-5-异丙基苯甲酸酯提供的阳离子相互作用与Lys890结合(图11C)。此外,还利用B16皮下移植模型研究了化合物9的体内活性。
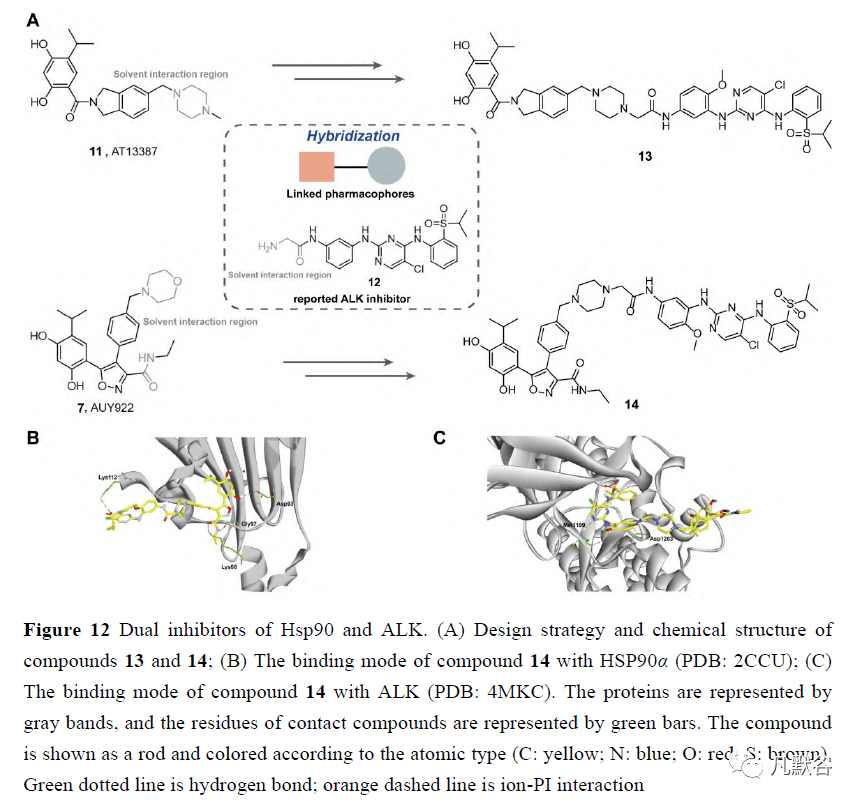
后来,本课题组设计和合成Hsp90/蛋白激酶多抑制剂,应用了一系列研究,包括通过流式细胞仪、蛋白质免疫印迹和蛋白质免疫沉淀,进行基于四甲基偶氮唑蓝的细胞增殖、细胞周期和凋亡分析,设计黑色素瘤异种移植模型,以及免疫组织化学/免疫荧光分析,以确定在先前的研究中开发的一种新型Hsp90/PI3K双重抑制剂DHP1808(10)的治疗效力和分子机制(图11A)。通过激活Fas/FasL信号通路诱导黑色素瘤细胞A375的细胞增殖抑制,并在Hsp90-EGFR相互作用和下游MAPK信号通路中断后诱导细胞周期停滞和细胞迁移或侵袭抑制。此外,DHP1808在肿瘤和正常组织中诱导的下垂程度低于Hsp90和PI3K抑制剂的组合,从而表明药物是安全的。 2.5 Hsp90和其他癌症相关靶点的双重抑制剂 ALK是分子伴侣Hsp90的客户蛋白之一,其成熟、激活和稳定性都受到Hsp90的调控[2]。Hsp90抑制剂的使用已被认为是克服ALK抑制剂诱导的耐药性的一种替代策略。Hsp90抑制剂的临床试验正在进行中,以评估它们与碱性磷酸酶抑制剂联合使用的抗癌效果[86]。2018年,Geng等[87]开发了新型Hsp90/ALK双靶向抑制剂,用于克服ALK抑制剂治疗引起的耐药性。在第二代ALK抑制剂12和Hsp90抑制剂AT13387(11)或AUY922(7)的基础上,他们设想间苯二酚连接的2, 4二氨基嘧啶在不同的连接点有不同的连接物,并可以作为同时靶向Hsp90和ALK的抑制剂(图12A)。结合生物学评价,将碱性磷酸酶抑制剂12的氨基片段引入到AT-13387的哌嗪部分和AUY922的异恶唑啉3-苯基的对位,合成了优化的化合物13和14。这两个化合物都显示出对ALK(9.8nmol·L -1 vs17.3nmol·L -1 )和Hsp90(40nmol·L -1 vs100nmol·L -1 )的高活性,证实了Hsp90对ALK和Akt的下调,H3122细胞中伴侣蛋白Hsp70的上调,以及对ALK成瘾的H3122细胞的高抗增殖作用(13nmol·L -1 vs11nmol·L -1 )。对接分析表明,14保留了几乎所有的关键氢键,类似于ALK抑制剂12与ALK的络合物和AUY922与Hsp90的络合物(图12B)。然而,由于哌嗪部分暴露在ALK激酶域的溶剂区,原始的ALK抑制剂部分暴露在Hsp90ATP结合口袋的溶剂区,并在Lys112和磺酰基之间形成氢键,导致14抗Hsp90的活性较低(图12C)。
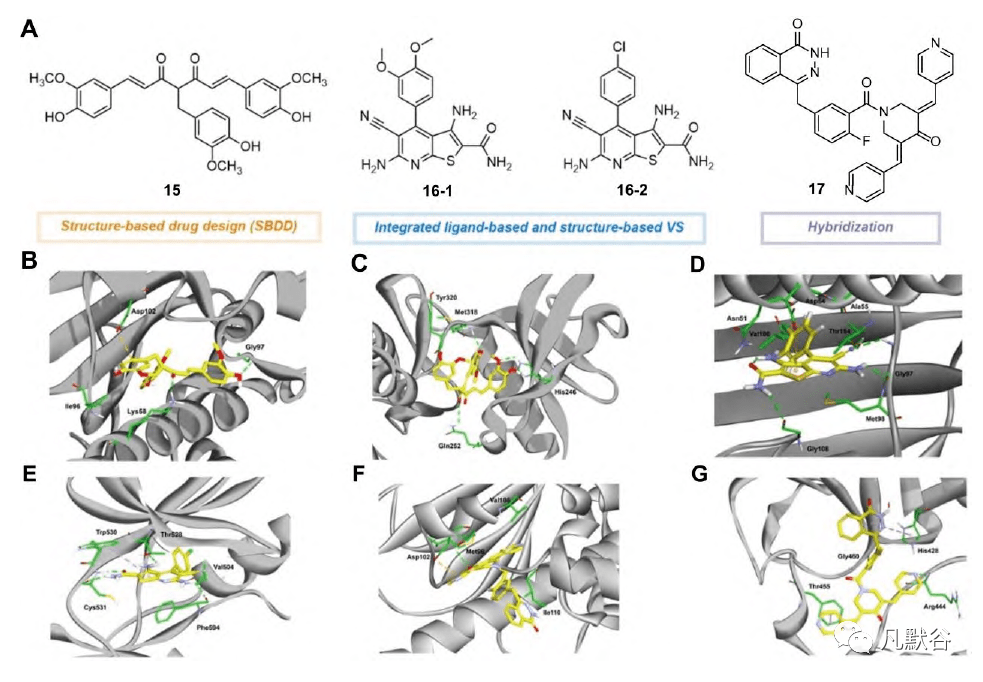
Gorre等[88]报道,BCR-ABL点突变株对Hsp90抑制剂格尔达那霉素(GA)和17-烯丙基氨基格尔德那霉素(17-AAG)敏感。2015年,Wu等[89]使用基于结构的药物设计,重点研究姜黄素类似物库,以寻找克服BCR-ABL突变和白血病干细胞引起的TKI耐药的抑制剂(图13A)。采用对接模型设计了一种合成姜黄素衍生物15,即C086。化合物15显示出比其母化合物姜黄素更好的活性和溶解性,并在小鼠SW480细胞移植模型中具有显著的抗增殖作用,且毒性相当低。它被认为是一种与Hsp90和BCR-ABL激酶结合的有效的新型抑制剂。一系列实验表明,C086抑制了结构域突变和过表达的WT ABL激酶活性。这些效应也是导致体外和慢性粒细胞白血病细胞对伊马替尼耐药的其他因素。C086与Hsp90物理结合,在体外影响其ATPase活性,破坏慢性髓系白血病(chronic myeloid leukemia, CML)细胞中Hsp90的伴侣功能,抑制人类白血病祖细胞或干细胞的生长,减少体内CML干细胞和祖细胞的生长,为治疗BCR-ABL诱导的白血病和其他对TKIs耐药的癌症类型提供了新的治疗策略。15与目标蛋白之间的分子对接研究如图13B、C所示。
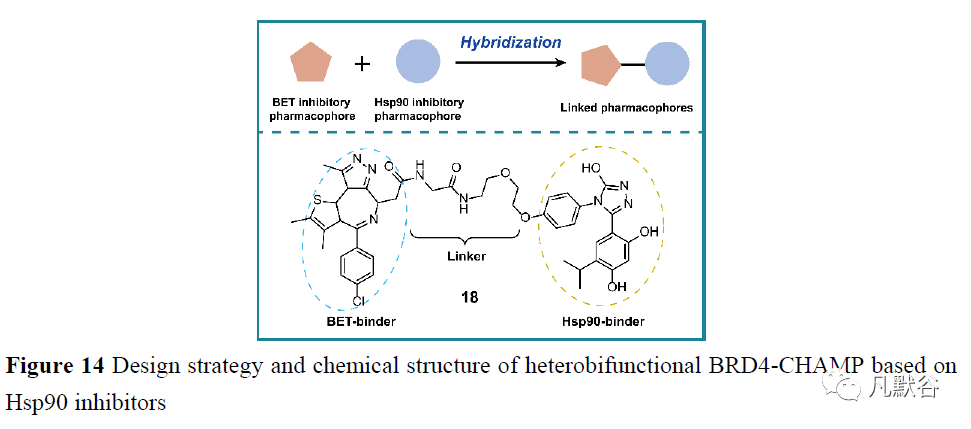
2017年,Anighoro等[90]设计并使用了一种结合配体和结构策略的VS方法,以确定两个创新化合物为Hsp90/B-Raf的双重抑制剂。第一个证据表明,Hsp90和B-Raf抑制剂共享部分重叠的化学空间。两个先导化合物16-1和16-2共享一个共同的硫代[2, 3-d]嘧啶骨架,在连接的苯环上有不同的取代(图13A)。这些化合物对所有受试靶标Hsp90、B-Raf,甚至对突变体B-RafV600E都显示出微摩尔活性。分子对接研究表明,这两种化合物都能与Hsp90的Asn251、Asp93和Thr184残基形成氢键网络。疏水作用是通过Hsp90残基Leu107和Phe138(图13D)在苯环上进行取代而形成的。在B-Raf中,这两种化合物都通过酰胺基和DFG基序残基附近的氰基和取代苯环与剩余的Cys532形成氢键(图13E)。 2020年,Lin等[91]采用杂交策略,将FDA批准的PARP-1抑制剂奥拉帕尼与他们之前合成的姜黄素衍生的Hsp90抑制剂C0817结合,获得了一些PARP和Hsp90的双靶点抑制剂。获得的分子表现出强烈的抗癌选择性细胞毒性(图13A)。受分子对接研究显示吡啶的氮原子可以与PARP-1中的残基Arg878形成氢键并与Hsp90形成键的启发,进行了2氟-5-[(4-氧代-3,4-二氢萘啶-1-基)甲基]苯甲酸和哌啶-4-酮的分子杂交,并在体外测试了生物活性。SAR分析表明,吸电子芳环,如吡啶环或卤素取代的苯环,对受试肿瘤细胞的抗增殖活性有较大贡献,而给电子基团,如甲氧基或羟基取代的苯环,导致抗癌活性的完全丧失。高效能的化合物17通过蛋白质印迹证实具有PARP抑制活性,并通过荧光淬灭进一步鉴定了Hsp90结合能力。化合物17可以下调乳腺癌1号基因(BRCA-1),Hsp90的客户蛋白之一和HR中的必需蛋白17和靶蛋白之间的分子对接研究如图13F、G所示。 2.6 针对Hsp90和其他癌症相关靶点的分子伴侣介导的蛋白质降解剂(chaperone-mediated protein degradation, CHAMP) 2021年,Foley等[92]开发了一种异双功能小分子化合物,即RNK05028(18),通过利用Hsp90结合部分独特的肿瘤选择性药代动力学来实现CHAMP。该化合物由3部分组成,一个目的蛋白的结合剂(BET-结合剂)、一个分子伴侣的结合剂(Hsp90-结合剂)和它们之间的连接物(图14)。它能在MV-4-11白血病细胞中形成BRD4:CHAMP:HSP90三元复合体,选择性降解依赖蛋白酶体的BRD4,抑制细胞增殖。
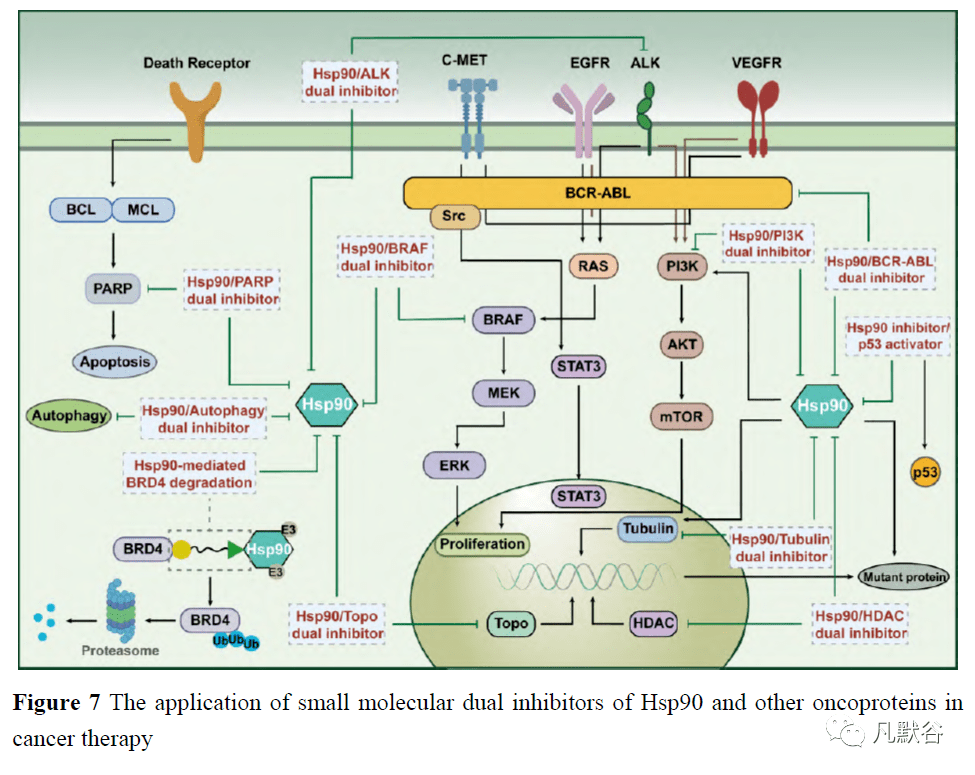
此外,CHAMP分子还可以降解非Hsp90蛋白,如突变的KRAS。体内试验表明,与血浆和正常组织相比,RNK05028在肿瘤中表现出更长的药代动力学,提高了安全边际。 3 结论 癌症这类复杂疾病会受到许多因素的影响,并且通常对单靶点治疗具有耐受性[93-95]。因此,多靶点疗法引起了研究复杂疾病的药物化学家的关注。由于联合疗法(即药物鸡尾酒)和伴随的药物相互作用风险,而多靶点药物具有明显的优势,多靶点配体的设计在过去10年中引起了巨大的科学兴趣[67, 68, 96]。虽然药物组合的双靶点或多靶点机制通常仅在临床成功的回顾性研究中披露,但双靶点或多靶点配体大多是根据联合治疗的临床数据、药物组合的表型筛选或硅晶入路的临床数据,通过合理的药物设计而获得的。 Hsp90主要被确定为癌症化疗的靶点,它的抑制剂是潜在的有效抗癌药物。在过去的几十年里,数十种Hsp90抑制剂已经达到了临床试验的不同阶段。其中,据资料显示口服Hsp90抑制剂pimitespib目前已在日本获批上市,用于治疗化疗后进展的胃肠道间质瘤;2023年2月6日该药获得中国临床试验审批,拟用于伊马替尼耐药的胃肠道间质瘤,pimitespib是一种值得关注和期待的抗癌治疗药物,期待该药未来能为更多肿瘤患者带来希望。因此,单药物分子对Hsp90等癌蛋白的双靶向/多靶向是开发新型抗癌药物的一种有力的途径。本文综述了Hsp90双抑制剂以及HDAC、tubulin、Topo II、PI3K、BCR-ABL、ALK、PARP等通路内或通路间肿瘤相关靶点的研究进展,包括杂交策略和VS方法在内的广泛研究致力于发现双Hsp90/HDAC抑制剂,与单Hsp90抑制剂和HDAC抑制剂的联合使用保持同步。也有人通过重叠药效团法和表型筛选等方法开发了Hsp90和微管蛋白的双重抑制剂,得到了两种化学实体,其中CDBT已通过体外和体内实验验证。虽然有文献重点介绍了双Hsp90/Topo II抑制剂,并建立了药效团模型,此外还有两种海洋甾萜类化合物被证明可以同时抑制Hsp90和Topo II,但目前还没有合成化学物质成功开发为新的双Hsp90/Topo II抑制剂。针对Hsp90和PI3K/Akt/mTOR的双抑制剂的设计和开发跟不上针对这些靶点的联合药物的速度。 综上所述,Hsp90和其他癌症相关靶点的双靶抑制剂目前显示出巨大的潜力,应进一步探索这些双靶抑制剂的治疗价值。由于Hsp90复合物在肿瘤组织中相对于正常组织处于高度活化状态,Hsp90结合小分子药效团在开发用于蛋白质降解的创新靶向技术CHAMP的异双功能小分子化合物中显示出独特的选择性药代动力学。认为Hsp90的双靶点抑制剂具有广阔的应用前景,但目前开发程度还很低。 尽管该领域已经使用了前瞻的计算方法,但还没有候选方法进入临床试验,这表明应该利用涉及药物化学、蛋白质组学、化学生物学、多药理学和计算化学的进一步综合方法。鉴于冷冻电子显微镜和AlphaFold人工智能预测等前沿技术正在推动结构生物学的快速发展,对癌蛋白结构的研究将有助于基于结构的双重抑制剂设计。在不久的将来,Hsp90的双重抑制剂有望在抗癌药物发现中发挥越来越重要的作用。 作者贡献 朱红萍、谢欣是本文的第一作者,负责文献资料的收集及文章的 撰写;覃蕊、黄维、刘燕青对文章修改提出建议,进行指导和帮助;彭成对本文进行了细致的修改;韩波为本文的通讯作者,负责提供撰写思路和内容框架组织等工作;何谷为本文的共同通讯作者,负责稿件修改完善等工作。 参考文献 详见 《药学学报》2023年 GastroPlus研讨班 · 2023 第十届生理药代动力学建模与模拟GastroPlus研讨班将采用线上+线下的方式举办(共5天:9月14-15号,9月21-23号) 本研讨班讲师将由美国Simulations Plus、上海凡默谷、GastroPlus资深用户的技术专家担任。 点击图片,查看详情 免责声明 本公众号发布的文章均为促进制药界同行的交流与学习;不用于任何商业用途。 我们尊重原创作品。选取的文章已明确注明来源和作者,版权归原作者所有,如涉及侵权或其他问题,请联系我们进行删除。 内容由凡默谷小编查阅文献选取,排版与编辑为原创。如转载,请尊重劳动成果, 注明【来源:凡默谷公众号】。 文章内容为作者观点,不代表本公众号立场。 文章搜索 本公众号开通往期文章任意搜功能啦 在公众号菜单栏中输入“搜索”,即可搜索往期微信内容 封面图片来源于:https://pixabay.com/zh/返回搜狐,查看更多 |

【本文地址】
今日新闻 |
点击排行 |
|
推荐新闻 |
图片新闻 |
|
专题文章 |