利用seqtk从基因组文件里面提取部分序列 |
您所在的位置:网站首页 › 怎么根据蛋白名称查基因序列数据 › 利用seqtk从基因组文件里面提取部分序列 |
利用seqtk从基因组文件里面提取部分序列
|
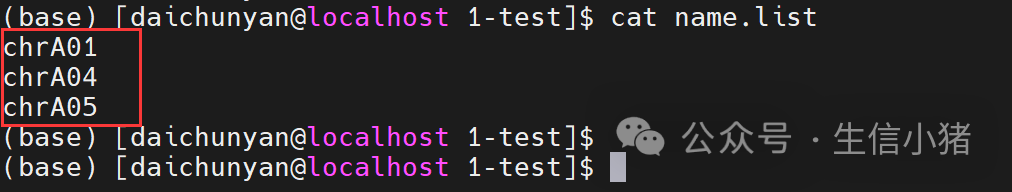
一、根据序列名提取固定序列 使用 seqtk subseq 命令从基因组文件里面提取部分序列 比如从下面文件里提取chrA01,chrA04,chrA05染色体的序列
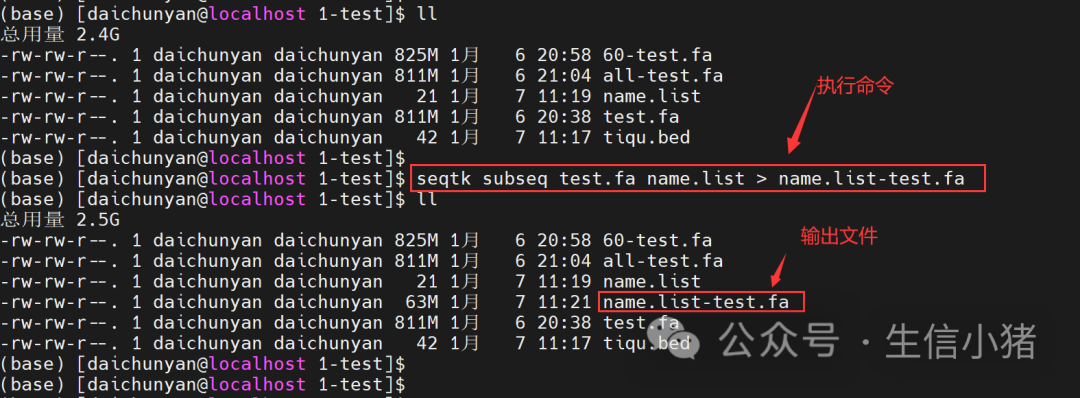
可以使用下面命令 seqtk subseq test.fa name.list > name.list-test.fa在这个命令里,name.list文件是自己整理的
解释命令: 1) seqtk subseq: 使用Seqtk工具的子命令,用于提取序列。 2) test.fa: 输入的FASTA格式文件,文件名为test.fa。 3) name.list: 染色体名称文件,用于指定要提取的序列。 4) tiqu-test.fa: 输出的FASTA格式文件,提取后的序列保存为name.list-test.fa。 在linux下的具体操作
看一下最终输出文件的结果,只包含想要提取的染色体序列
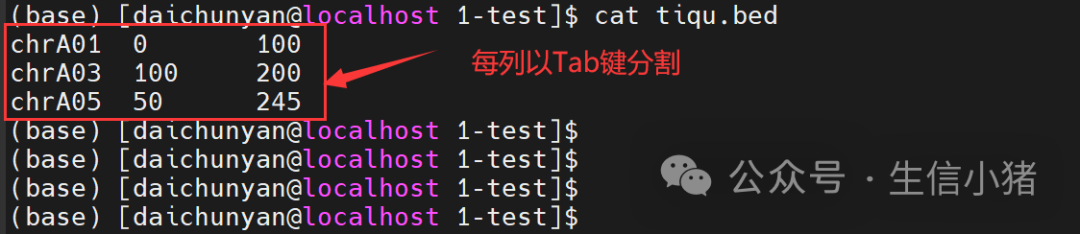
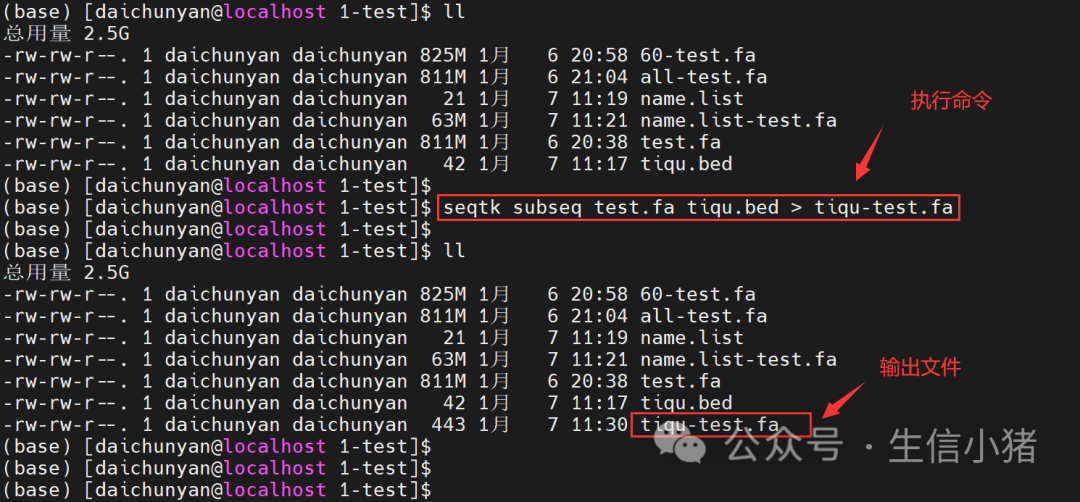
二、根据序列名和区域提取固定区域序列 可以使用下面命令 seqtk subseq test.fa tiqu.bed > tiqu-test.fa在这个命令里,tiqu.bed文件是自己整理的
tiqu.bed文件总共有3列,每列以Tab键分割,第1列是染色体名称,第2列是想要截取染色体上的起始位置,第3列是截取染色体上的结束位置。这里要注意,染色体第1个碱基位置的索引是0,即如果你想要截取某条染色体1到100位置的碱基,那么在bed文件里就要写成0到100。 在linux下的具体操作
看一下最终输出文件的结果,只包含想要提取的固定区域序列
|

【本文地址】
公司简介
联系我们
今日新闻 |
点击排行 |
|
推荐新闻 |
图片新闻 |
|
专题文章 |