SusMat.:理论计算P对锰和氮掺杂石墨烯催化剂促氧还原 |
您所在的位置:网站首页 › 原子轨道3dz2 › SusMat.:理论计算P对锰和氮掺杂石墨烯催化剂促氧还原 |
SusMat.:理论计算P对锰和氮掺杂石墨烯催化剂促氧还原
|
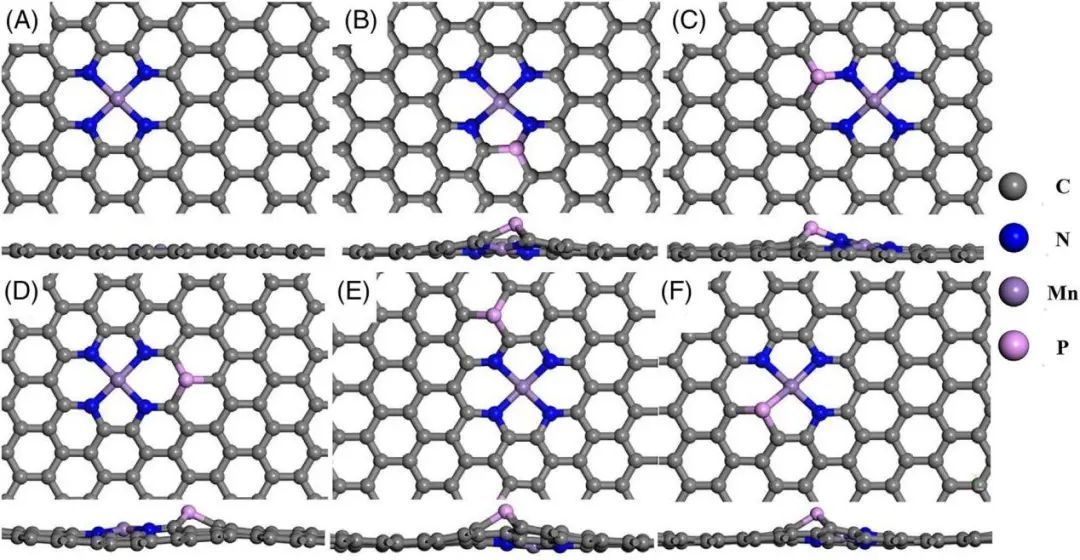
成果简介 近年来,新型能源转换装置(燃料电池和锌-空气电池)的探索得到了广泛的关注。开发促进氧还原反应(ORR)的有效催化剂是解决ORR反应缓慢动力学问题的关键,最终实现这些装置的商业化。铂及铂基合金被认为是高效稳定的ORR电催化剂,然而,贵金属铂的高成本限制了这些新型能源转换装置的大规模应用,所以开发非贵金属催化剂,实现新能源装置的广泛应用具备重要的研究意义。 大连理工大学刘安敏,美国纽约州立大学武刚和哈尔滨工业大学安茂忠等人基于密度泛函理论计算,对非贵金属锰配位N、P共掺杂石墨烯材料进行了ORR的理论研究,希望可以获得具有优异氧还原反应活性的MnN4催化剂,本研究可为合理设计高性能锰基碳材料电催化剂提供理论指导。 计算方法 所有DFT计算均使用维也纳从头算模拟包(VASP)进行建模,交换相关能通过广义梯度近似下(GGA)的Perdew – Burke – Ernzerhof交换相关函数进行理论计算。作者采用投影增强波赝势(PAW)方法进行离子-电子相互作用的研究,截断能设置为500 eV,结构优化的力收敛极限为0.02 eV/Å,采用DFT+U方法矫正Mn元素能量,U设置为5 eV。 结果与分析 对六种不同的催化剂结构MnN4-G、MnN4-P1-G、MnN4-P2-G、MnN4-P3-G、MnN4-P4-G、MnN3P-G进行优化,优化结构如图1所示。通过计算体系形成能可以发现,除了MnN4-P3-G之外,所有催化剂的生成能都在−2.697和−2.135eV范围内,说明所有结构都具有良好的热力学稳定性。
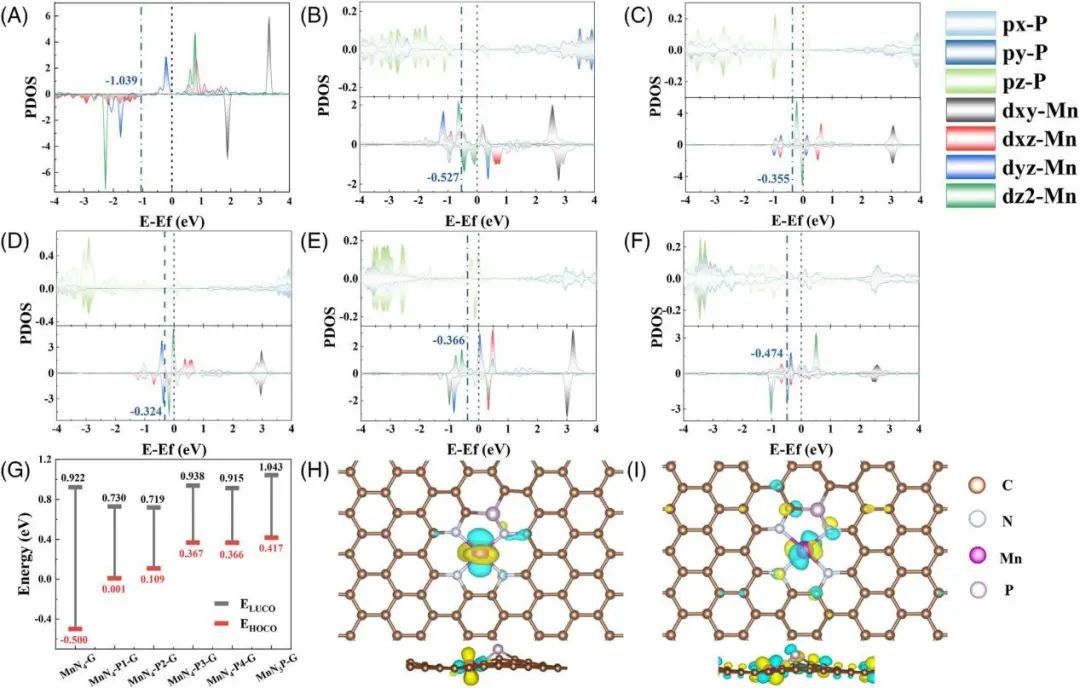
图1 催化剂模型 态密度(DOS)和d带中心被广泛用于电子结构分析,为ORR的催化活性提供支持。在Mn-N-C催化剂中引入P原子导致电子结构根据部分和全部DOS发生变化(图2)。从图2可以看出,P原子的3p轨道和Mn原子的3d轨道之间发生了强烈的杂化。 费米能级附近的DOS表明,MnN4-P1-G和MnN4-P2-G催化剂中Mn原子的三维轨道(3dz2和3dyz)与P原子的3pz轨道存在重叠,而MnN4-P3-G催化剂中Mn原子的3dxz和3dz2轨道与P原子的3pz轨道存在较大的电子重叠。同样,MnN4-P4-G中Mn原子的3dyz、3dz2和P原子的pz之间也存在电子重叠。 然而,轨道中心越贴近于费米能级越会导致含O中间体的吸附更强,这可能会由于更高的能垒而对催化活性产生不利影响,相比之下,MnN4-P1-G、MnN4-P4-G和MnN3P-G三种催化剂的轨道中心相对适中(介于0.2317 ~ 0.9465之间),有利于ORR的提高。还可以得出结论,P原子的引入会导致d带中心发生正位移。
图2 催化剂电子结构 同时,作者对催化剂对于OER中间体的吸附能ΔE进行了排名,排名MnN4-P3-G |

【本文地址】
今日新闻 |
点击排行 |
|
推荐新闻 |
图片新闻 |
|
专题文章 |