| 【蛋白结构AI预测时代】在Colab上使用AlphaFold2教程 | 您所在的位置:网站首页 › 谷歌的alphafold › 【蛋白结构AI预测时代】在Colab上使用AlphaFold2教程 |
【蛋白结构AI预测时代】在Colab上使用AlphaFold2教程
|
Alphafold2 开源了!!!这将进一步推动学界预测和设计蛋白。
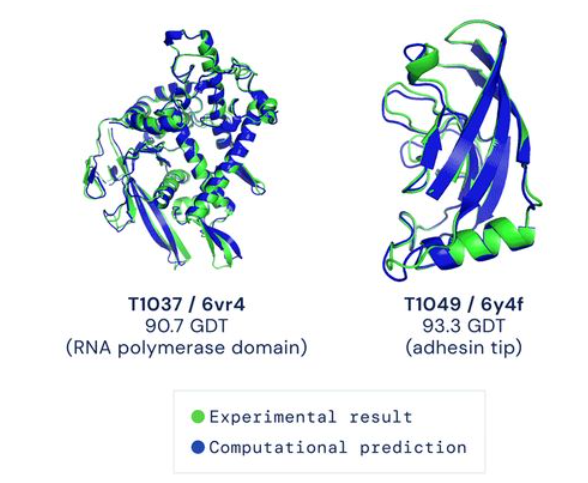
可以看到官网上给出的结果图,结构生物学实验解得的结构与预测的别无二致。 链接:https://colab.research.google.com/github/sokrypton/ColabFold/blob/main/AlphaFold2.ipynb#scrollTo=UGUBLzB3C6WN —————————分割线下的可以不用看啦(老)———————————— 但模型的数据与预测所需资源过大,跑起来也比较费时,这里用Sergey Ovchinnikov 提供的“alphafold_single_sequence.ipynb”的代码,快速跑通看一下它的效果。 先是导入和安装各种库 %%bash git clone https://github.com/deepmind/alphafold.git mv alphafold alphafold_ mv alphafold_/alphafold . %%bash wget -qnc https://storage.googleapis.com/alphafold/alphafold_params_2021-07-14.tar tar -xf alphafold_params_2021-07-14.tar rm alphafold_params_2021-07-14.tar mkdir params mv params_* params/ %%bash pip -q install biopython pip -q install dm-haiku pip -q install ml-collections pip -q install mock pip -q install py3Dmol from typing import Dict import os import mock import numpy as np import pickle import py3Dmol from alphafold.common import protein from alphafold.data import pipeline from alphafold.data import templates from alphafold.model import data from alphafold.model import config from alphafold.model import model定义一个结构预测函数。 def predict_structure( fasta_path: str, fasta_name: str, output_dir_base: str, data_pipeline: pipeline.DataPipeline, model_runners: Dict[str, model.RunModel], random_seed: int): """Predicts structure using AlphaFold for the given sequence.""" output_dir = os.path.join(output_dir_base, fasta_name) if not os.path.exists(output_dir): os.makedirs(output_dir) msa_output_dir = os.path.join(output_dir, 'msas') if not os.path.exists(msa_output_dir): os.makedirs(msa_output_dir) # Get features. feature_dict = data_pipeline.process(input_fasta_path=fasta_path, msa_output_dir=msa_output_dir) # Run the models. for model_name, model_runner in model_runners.items(): processed_feature_dict = model_runner.process_features(feature_dict, random_seed=random_seed) prediction_result = model_runner.predict(processed_feature_dict) unrelaxed_protein = protein.from_prediction(processed_feature_dict,prediction_result) unrelaxed_pdb_path = os.path.join(output_dir, f'unrelaxed_{model_name}.pdb') with open(unrelaxed_pdb_path, 'w') as f: f.write(protein.to_pdb(unrelaxed_protein))将query_sequence更改为你自己的序列。 # CHANGE THIS LINE TO YOUR FAVE SEQUENCE! :D query_sequence = "GWSTELEKHREELKEFLKKEGITNVEIRIDNGRLEVRVEGGTERLKRFLEELRQKLEKKGYTVDIKIE" # fake template output_templates_sequence = [] output_confidence_scores = [] templates_all_atom_positions = [] templates_all_atom_masks = [] for _ in query_sequence: templates_all_atom_positions.append( np.zeros((templates.residue_constants.atom_type_num, 3))) templates_all_atom_masks.append(np.zeros(templates.residue_constants.atom_type_num)) output_templates_sequence.append('-') output_confidence_scores.append(-1) output_templates_sequence = ''.join(output_templates_sequence) templates_aatype = templates.residue_constants.sequence_to_onehot(output_templates_sequence, templates.residue_constants.HHBLITS_AA_TO_ID) template_features = {'template_all_atom_positions': np.array(templates_all_atom_positions)[None], 'template_all_atom_masks': np.array(templates_all_atom_masks)[None], 'template_sequence': [f'none'.encode()], 'template_aatype': np.array(templates_aatype)[None], 'template_confidence_scores': np.array(output_confidence_scores)[None], 'template_domain_names': [f'none'.encode()], 'template_release_date': [f'none'.encode()]} # fake pipeline for testing data_pipeline_mock = mock.Mock() data_pipeline_mock.process.return_value = { **pipeline.make_sequence_features(sequence=query_sequence, description="none", num_res=len(query_sequence)), **pipeline.make_msa_features(msas=[[query_sequence]], deletion_matrices=[[[0]*len(query_sequence)]]), **template_features } fasta_path = os.path.join('target.fasta') with open(fasta_path, 'wt') as f: f.write(f">A\n{query_sequence}") fasta_name = 'none' out_dir = "." # load model_1 model_runners = {} model_name = "model_1" model_config = config.model_config(model_name) model_config.data.eval.num_ensemble = 1 model_params = data.get_model_haiku_params( model_name=model_name, data_dir=".") model_runner = model.RunModel(model_config, model_params) model_runners[model_name] = model_runner预测结构,这个蛋白示例约有68个氨基酸,耗时约2分钟。 predict_structure( fasta_path=fasta_path, fasta_name=fasta_name, output_dir_base=".", data_pipeline=data_pipeline_mock, model_runners=model_runners, random_seed=0)查看结构,并可以在文件栏下载pdb格式的蛋白文件。 p = py3Dmol.view(js='https://3dmol.org/build/3Dmol.js') p.addModel(open("none/unrelaxed_model_1.pdb",'r').read(),'pdb') p.setStyle({'cartoon': {'color':'spectrum'}}) p.zoomTo() p.show()
|
 ——————————— 最新最省力的方法———————————————— 直接点击Colab的Alphafold2预测链接,输入序列,全部运行就可以预测了。
——————————— 最新最省力的方法———————————————— 直接点击Colab的Alphafold2预测链接,输入序列,全部运行就可以预测了。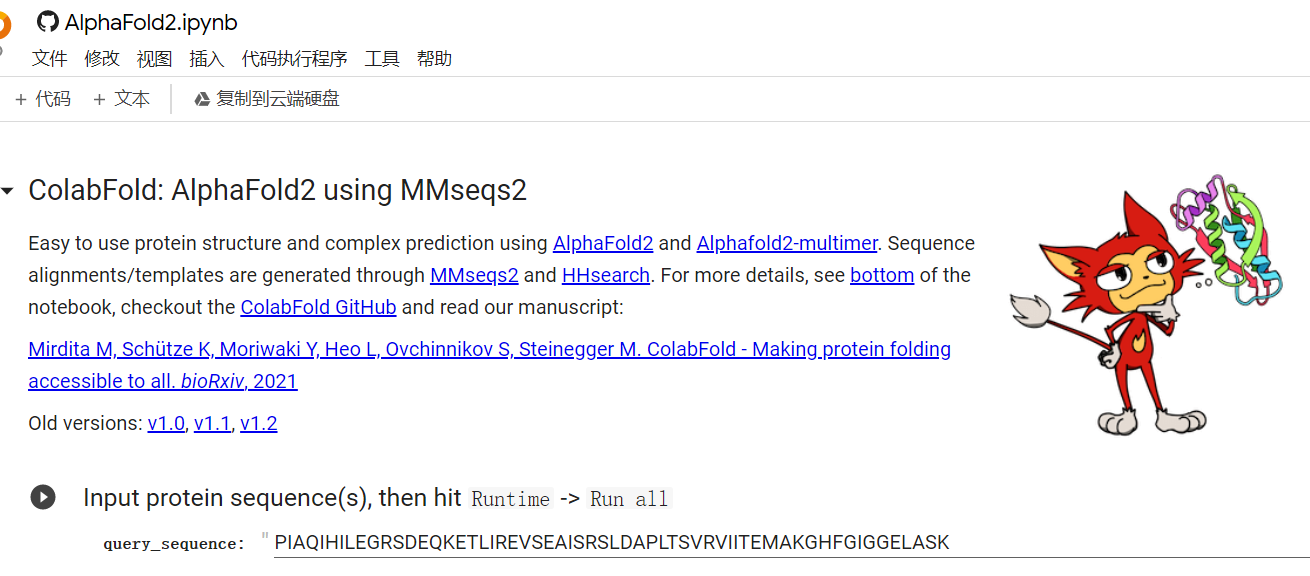
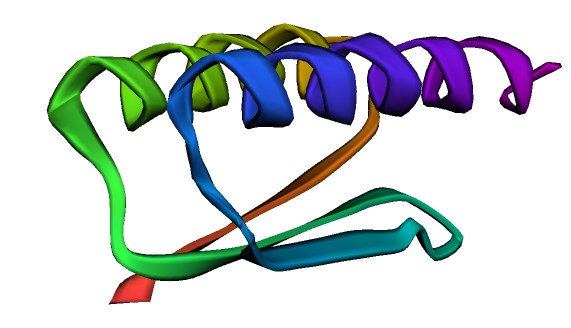
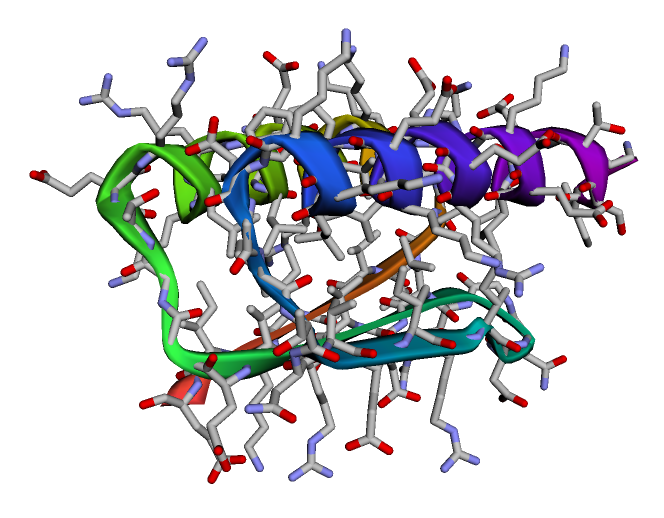
【本文地址】
公司简介
联系我们