| 肥厚型心肌病 | 您所在的位置:网站首页 › 肥厚性心肌病的症状是怎样的 › 肥厚型心肌病 |
肥厚型心肌病
|
肥厚型心肌病(HCM)是一种以心肌肥厚为特征的最常见遗传性心脏病,是导致青少年和运动员猝死的主要原因之一。肥厚型心肌病的发生率是1/500,随着诊断技术的进步,现在研究发现发生率甚至高达1/200。本文根据2022年中国肥厚型心肌病指南、2020年美国心脏协会AHA/美国心脏病学会ACC肥厚型心肌病诊疗指南和2014年欧洲心血管病协会ESC肥厚型心肌病诊治指南,结合最新研究,和大家介绍一下HCM的病因和诊治。 1. 肥厚型心肌病的病因据数据显示,约50%的HCM患者具有家族遗传性。目前HCM的分子遗传学研究证实此病为肌小节结构蛋白的编码基因突变,其中MYBPC3基因和MYH7基因是HCM患者最常见的两种致病基因。多数由基因变异引起的HCM的遗传模式为常染色体显性遗传模式,每个受累家系成员的后代都有50% 的概率会遗传到这种致病基因。但由于致病基因变异存在外显不全及年龄依赖的表达,携带致病基因不一定出现临床表型。部分HCM 患者可能携带单个基因的多个变异(复合变异)或≥2个相同或不同基因的杂合变异,统称为复杂基因变异现象。研究发现,携带复杂基因变异者发病更早,临床表型更重,预后更差。但基因突变引起的HCM发病机制仍不明确。有研究者推测基因突变导致肌纤维收缩功能受损,从而代偿性的出现心肌肥厚和舒张功能障碍;也有研究者提出基因突变导致钙循环或钙敏感性受扰,能量代谢受到影响,从而出现心肌肥厚、纤维化、肌纤维排列紊乱及舒张功能改变。这些学说虽然互为补充地解释了HCM的致病机制,但均难以完全阐明。临床诊断HCM时,需要和其他心肌肥厚的病因做鉴别诊断。除了高血压、主动脉瓣狭窄、心肌炎和运动员心脏外,心肌肥厚40-60%是肥厚型心肌病,5-10%的心肌肥厚是由其他遗传性或非遗传性疾病引起,包括先天性代谢性疾病(如糖原贮积病、肉碱代谢疾病、溶酶体贮积病),神经肌肉疾病(如Friedreich共济失调),线粒体疾病,畸形综合征,系统性淀粉样变等,这类疾病临床罕见或少见。另外还有25-30%的心肌肥厚原因不明。 2. 肥厚型心肌病的病理生理HCM病理生理机制复杂,主要包括左心室流出道梗阻(LVOTO)、二尖瓣反流(MR)、舒张功能不全、心肌缺血和自主神经功能不全等。在临床中,对于一个HCM患者,可能以一种机制为主,也可能同时存在相互作用的多种机制。以下为HCM各种病理生理机制产生的主要原因:2.1产生LVOTO 的两种机制:①非对称性室间隔肥厚,尤其是室间隔基底部增厚及二尖瓣解剖结构改变(包括瓣叶拉长、乳头肌前移)造成LVOT 狭窄引起的机械性梗阻;②二尖瓣前叶收缩期前移现象加重LVOT 狭窄引起的动力性梗阻。2.2产生MR原因主要有两方面:①继发于LVOTO ;②原发性或内在性的二尖瓣相关结构异常,如二尖瓣叶过度拉长、异常乳头肌插入、乳头肌前向移位等。2.3产生舒张功能不全机制主要包括两方面:①心肌缺血、缺氧、能量代谢障碍,导致心肌细胞内舒张期钙再摄取异常,导致心肌主动松弛能力受损;②心室肥厚、心肌纤维化、心室几何形状改变等,导致心室壁顺应性降低(僵硬度增加),心室被动充盈受限。2.4产生心肌缺血原因包括四个方面:①心肌细胞肥大,导致心肌的氧供和需求失衡;②心室壁内小冠状动脉管壁增厚,管腔狭窄,血管分布密度降低;③冠状动脉微血管功能障碍导致冠状动脉血流储备降低;④部分患者可以合并冠状动脉肌桥及冠状动脉粥样硬化病变。 3.肥厚型心肌病的类型HCM的分型主要依据患者的血流动力学、心脏肥厚部位及遗传学规律。其分型如下图所示,不同分型的患者,其治疗方式及预后情况也不同。3.1 根据血流动力学分型:是临床最常用的HCM分型方法,有利于指导治疗措施的选择。3.1.1梗阻性HCM:根据梗阻部位又可以分为左室流出道梗阻(LVOTO)、左心室中部梗阻及左心室心尖部梗阻。通常所说的梗阻性HCM 是指LVOTO,即LVOT 瞬时峰值压差≥30mmHg;同时可以分为静息梗阻性(静息状态存在LVOTO)和隐匿梗阻性(静息无梗阻,激发试验时出现LVOTO)。3.1.2非梗阻性HCM:指静息时和激发时LVOT 峰值压差均2.5 时可诊断。(2)对于有明确HCM 家族史或者致病基因检测阳性的儿童,建议采用z值>2的界值。 5.肥厚型心肌病的合并症和并发症5.1心房颤动:是HCM患者最常见的心律失常之一,约18-20%的HCM患者合并房颤,增加患者卒中和周围栓塞的风险,明显影响患者的生活质量。5.2心力衰竭:通常由左心室舒张功能异常引起,表现为射血分数保留的心力衰竭(HFpEF)。少数患者进展至终末期HCM,出现严重的左心室收缩功能障碍,表现为射血分数降低的心力衰竭(HFrEF)。HCM患者发生心衰时出现呼吸困难症状,发生率约15%。5.3左室心尖部室壁瘤:是HCM相对少见但是危险的并发症,发生率在1-5% 之间,多见于心尖肥厚型心肌病和左心室中部梗阻性肥厚型心肌病等特殊类型的HCM。左室心尖部室壁瘤是HCM患者发生心脏性猝死的危险因素之一,与HCM的不良预后密切相关,需要植入ICD。5.4心腔内血栓:根据心腔内血栓部位可以分为心房血栓和心室血栓。心房血栓(包括心耳血栓)主要与房颤相关。左心室血栓在HCM患者中相对罕见,可见于心脏室壁瘤和终末期重症患者。6.肥厚型心肌病的治疗 HCM的治疗目标包括缓解临床症状,改善心脏功能,延缓疾病进展,减少疾病死亡。治疗原则包括:1. 无症状HCM 患者需要定期进行临床评估。2. 对于有症状的非梗阻性HCM 患者,主要针对合并症和并发症进行治疗。3.对于有症状的梗阻性HCM 患者,使用药物治疗或手术治疗方式改善症状。4. 所有HCM患者都应常规开展心脏性猝死的风险评估和危险分层,进行相应预防和治疗。6.1药物治疗的主要目的是缓解HCM 患者的症状,目前尚没有证据显示药物治疗可以改变HCM的自然病史。症状性梗阻性HCM 患者的药物治疗种类主要包括以下五种:① β受体阻滞剂:是最早被研究用于治疗HCM患者的药物,可以抑制心肌收缩力,降低左室流出道(LVOT)压差,减轻左室流出道梗阻(LVOTO);可以减慢心率,改善心室舒张期充盈,明显改善患者的心功能和生活质量。目前,β受体阻滞剂多作为一线治疗药物,儿童HCM 患者也可以使用β受体阻滞剂。常用的是美托洛尔和比索洛尔。② 非二氢吡啶类钙离子通道阻断剂(CCB):具有负性肌力和负性频率作用,可以减轻LVOTO,改善舒张期心室充盈程度和患者症状。对于β受体阻滞剂治疗无效、无法耐受或有禁忌的症状性梗阻性HCM 患者,推荐使用非二氢吡啶类CCB,包括维拉帕米和地尔硫卓。③ 心肌肌球蛋白抑制剂:Mavacamten是选择性心肌肌球蛋白变构抑制剂,通过选择性降低心肌肌球蛋白重链的ATP 酶活性,可逆地抑制肌球蛋白-肌动蛋白横桥的过量形成,同时促使整个肌球蛋白群体转向节能的超松弛状态,从而抑制心肌过度收缩、改善舒张顺应性及能量代谢。Aficamten是一种新型选择性小分子心肌肌球蛋白抑制剂。目前临床试验表明这一类药物能显著降低左室流出道压力差,缓解梗阻,显著减少患者临床手术的必要性,笔者认为是非常有临床前景的一类药物,在中国也很快会上市。④ 丙吡胺和西苯唑啉:都属于抗心律失常药物,可以降低LVOT压差。对于使用β受体阻滞剂和非二氢吡啶类CCB后仍有与LVOTO相关症状的重症患者,可考虑使用。因为可能潜在都致心律失常作用,实际上目前在中国临床很少应用。6.2手术治疗包括室间隔心肌消融术(SMA)和外科室间隔心肌切除术(SSM),可统称为室间隔减容术(SRT)。SMA包括用酒精间隔消融术(ASA)、经皮心肌内间隔射频消融术(PIMSRA,又称Liwen术式)、心内膜间隔射频消融术(ERASH)等。其中Liwen术式是由中国西京医院刘丽文教授首创,对既往术式进行很好的革新和改进,目前正被越来越多的国家接受,是国人的骄傲。室间隔心肌消融术外科室间隔心肌切除术 手术适应证:(1)临床标准:患者经充分药物治疗后仍然存在严重呼吸困难或胸痛症状(NYHA心功能Ⅲ级或Ⅳ级),或者存在其他与活动相关的症状(如反复发生晕厥或近乎晕厥),与LVOTO 相关,影响患者日常活动和生活质量。(2)血流动力学标准:静息或激发时LVOT峰值压差≥50mmHg,与室间隔肥厚或二尖瓣SAM 现象相关。(3)解剖标准:由术者个人判断拟行手术的室间隔厚度是否能足够安全有效地进行操作。对于无症状且活动耐力正常的梗阻性HCM患者或者虽然有症状但经优化药物治疗可以控制,则不推荐进行SRT手术。 7.危险评估和猝死的预防肥厚型心肌病是患者,特别是青少年患者猝死的主要原因,一旦发生,对患者及家庭的危害极大,因此如何正确评估肥厚型心肌病患者猝死的风险以及预防至关重要,笔者特另辟一文进行解释,请详见《肥厚性心肌病患者如何预防猝死》一文。 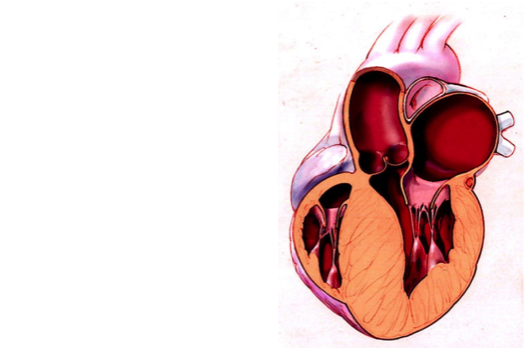
|
【本文地址】