| 一种甘油改性的聚乙烯醇水凝胶及其制备方法和应用 | 您所在的位置:网站首页 › 聚乙烯醇的改性方法 › 一种甘油改性的聚乙烯醇水凝胶及其制备方法和应用 |
一种甘油改性的聚乙烯醇水凝胶及其制备方法和应用
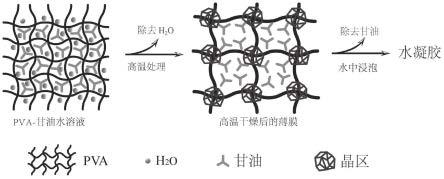 1.本发明属于水凝胶薄膜材料技术领域,具体涉及甘油改性的聚乙烯醇水凝胶及其制备方法和应用。 背景技术: 2.水凝胶是具有三维交联网络结构的典型“软湿”材料,可以在吸收大量水的同时仍保持其固体形状。水凝胶可以由合成聚合物如聚乙烯醇、聚丙烯酰胺、聚丙烯酸等进行制备,或者由天然聚合物,如纤维素、壳聚糖和淀粉进行制备。水凝胶在生物,食品以及电子器件等领域应用广泛,但是由于大部分水凝胶制备工艺复杂,力学性质差,严重限制了水凝胶的应用,因此水凝胶的力学性能提升的制备工艺一直是科学与工业界十分关注的研究热点。3.聚乙烯醇(pva)是一种生物可降解、非致癌、无毒、生物相容性、水溶性和廉价的聚合物,具有良好的物理性能,如良好的透明度、低界面张力和高溶胀率,是常用的水凝胶基质。大部分研究通过冻融的方法,获得物理交联的pva水凝胶。但是,pva水凝胶却存在着机械强度差的问题(《15mpa),而且冻融方法耗能多,时间长,不利于大规模的工业化生产。 技术实现要素: 4.本发明的目的在于提供一种绿色,简单,低成本地制备高力学性能pva水凝胶的方法。5.为达到上述目的,本发明采用以下技术方案:6.利用甘油对pva网络结构与晶区形态和数量进行调节,并利用高温条件下高效除水和非酶促焦糖化作用来增加分子间的相互作用力,从而改善pva水凝胶的力学性质。7.根据本发明具体实施方式的甘油改性的聚乙烯醇水凝胶,所述聚乙烯醇水凝胶由包括以下步骤的方法制备而成:8.(1)取聚乙烯醇基质、甘油分别加入水中,加热,搅拌溶解,得到混合溶液,9.(2)将步骤(1)得到的混合溶液流延成膜,干燥,得到薄膜;10.(3)将步骤(2)得到的薄膜在110~140℃、真空度为0~0.3atm条件下,干燥;11.(4)将步骤(3)干燥后的薄膜置于去离子水中,浸泡,得到聚乙烯醇水凝胶。12.根据本发明具体实施方式的甘油改性的聚乙烯醇水凝胶,步骤(1)中,将聚乙烯醇基质、甘油分别加入水中后,在110~130℃加热条件下加热10~30分钟,再以500~800r/min的速度,65~85℃搅拌至颗粒完全溶解,并超声去除气泡。13.根据本发明具体实施方式的甘油改性的聚乙烯醇水凝胶,步骤(1)中,原料的用量分别为:相对于水,聚乙烯醇基质质量含量4~10wt%、甘油体积百分含量2~5%。14.根据本发明具体实施方式的甘油改性的聚乙烯醇水凝胶,步骤(3)中,将薄膜置于真空干燥箱中120℃,真空度0-0.1atm条件下,干燥12h。15.根据本发明具体实施方式的甘油改性的聚乙烯醇水凝胶的制备方法,包括以下步骤:16.(1)取聚乙烯醇基质、甘油分别加入水中,加热,搅拌溶解,得到混合溶液;17.(2)将步骤(1)得到的混合溶液流延成膜,干燥,得到薄膜;18.(3)将步骤(2)得到的薄膜在110~140℃、真空度为0~0.3atm条件下,干燥;19.(4)将步骤(3)干燥后的薄膜置于去离子水中,浸泡,得到聚乙烯醇水凝胶。20.根据本发明具体实施方式的甘油改性的聚乙烯醇水凝胶的制备方法,步骤(1)中,将聚乙烯醇基质、甘油分别加入水中后,在110~130℃加热条件下加热10~30分钟,再以500~800r/min的速度,65~85℃搅拌至颗粒完全溶解,并超声去除气泡。21.步骤(1)中,原料的用量分别为:相对于去离子水基质质量百分含量为4~10wt%、甘油体积百分含量为1~5wt%。22.本发明的再一目的在于提供一种“掺杂”(壳聚糖)的甘油改性的聚乙烯醇水凝胶。23.根据本发明具体实施方式的甘油改性的聚乙烯醇水凝胶,所述聚乙烯醇水凝胶由包括以下步骤的方法制备而成:24.(1)取聚乙烯醇基质、甘油和壳聚糖分别加入水中,加热,搅拌溶解,得到混合溶液;25.(2)将步骤(1)得到的混合溶液流延成膜,干燥,得到薄膜;26.(3)将步骤(2)得到的薄膜在110~140℃、真空度为0~0.3atm条件下,干燥;27.(4)将步骤(3)干燥后的薄膜置于去离子水中,浸泡,得到聚乙烯醇水凝胶。28.根据本发明具体实施方式的甘油改性的聚乙烯醇水凝胶,步骤(1)中,将聚乙烯醇基质、甘油、壳聚糖分别加入水中后,在110~130℃加热条件下加热10~30分钟,再以500~800r/min的速度,65~85℃搅拌至颗粒完全溶解,并超声去除气泡。29.根据本发明具体实施方式的甘油改性的聚乙烯醇水凝胶,步骤(1)中,相对于水,聚乙烯醇基质质量含量4~10wt%、甘油体积百分含量2~5%,壳聚糖的质量含量0.1~0.2wt%。30.根据本发明具体实施方式的甘油改性的聚乙烯醇水凝胶,步骤(1)中,向水中加入0.3-1wt%的助溶剂。助溶剂可以选择醋酸,同时,本发明还可以采用柠檬酸,草酸、琥珀酸、苹果酸、己二酸。31.根据本发明具体实施方式的甘油改性的掺杂聚乙烯醇水凝胶,步骤(1)中,向水中加入改性生物高分子壳聚糖基质,也可以用蛋白,纤维素,淀粉,琼脂糖,魔芋胶来代替。32.根据本发明具体实施方式的甘油改性的聚乙烯醇水凝胶,步骤(3)中,将薄膜置于真空干燥箱中120℃,真空度0~0.1atm条件下,干燥12h。33.本发明方法制备得到的甘油改性的聚乙烯醇水凝胶可应用于生物医药、食品以及电学器件等领域中。34.本发明的有益效果:35.本发明采用甘油作为改性剂,调节聚乙烯醇水凝胶内部交联结构,形成有效增强分子间作用力的网络结构,显著提高了水凝胶的力学性能,改善抗水性能,其中以单纯聚乙烯醇作为基质的水凝胶其拉伸强度可达26.31±0.27mpa,溶胀率56.44±2.01%,掺入壳聚糖的聚乙烯醇为基质的水凝胶,其拉伸强度可达27.99±1.40mpa,溶胀率为81.18±4.42%。36.本发明的方法没有采用传统的高能耗,难大批量制备的冻融方式,而是通过加热与甘油调节的方法去制备pva水凝胶,降低了能耗,节省了时间,适合工业化生产。制备方法简单,得到的水凝胶具有优异的力学性质以及抗水性能能,进一步拓宽pva水凝胶的应用范围与潜力。附图说明37.为了更清楚地说明本发明实施例或现有技术中的技术方案,下面将对实施例或现有技术描述中所需要使用的附图作简单地介绍,显而易见地,下面描述中的附图仅仅是本发明的一些实施例。38.图1为本发明的聚乙烯醇水凝胶的机理示意图;39.图2显示不同甘油添加量的pva6-glx水凝胶薄膜的扫描电镜的微观结构图,其中,pva6-gl0(a),pva6-gl1(b),pva6-gl2(c),pva6-gl3(d),pva6-gl4(e);40.图3是pc-glx水凝胶薄膜循环拉力图,(a)pc-gl2,(b)pc-gl3,(c)pc-gl4;41.图4显示pc-glx水凝胶薄膜sem图像,pc-gl0(a),pc-gl1(b),pc-gl2(c),pc-gl3(d),pc-gl4(e),pc-gl5(f)。具体实施方式42.为使本发明的目的、技术方案和优点更加清楚,下面将对本发明的技术方案进行详细的描述。显然,所描述的实施例仅仅是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动的前提下所得到的所有其它实施方式,都属于本发明所保护的范围。43.本发明的反应原理如下:取聚乙烯醇基质、甘油分别加入水中,加热,搅拌溶解,得到pva-甘油混合溶液,将该混合溶液流延成膜,高温干燥,离去水分,得到干燥的薄膜。干燥后的薄膜置于去离子水中,浸泡,除去甘油即可得到聚乙烯醇水凝胶。44.本方法通过改变水凝胶配方中的甘油用量可以调节pva的网络结构与晶区形态和数量,利用高温条件可以高效离去水分,同时高温诱导的非酶促焦糖化作用可以促进pva分子间氢键的形成,增强分子间的相互作用力,如图1所示。因此甘油的用量和高温干燥温度可以调控pva水凝胶的微观结晶状态,从而显著提升pva水凝胶的力学性质,改善抗水性能。45.下面通过具体的实施例对发明进行说明。46.为简便起见,本发明实施例采用缩写符号pvax-gly来指定单纯使用甘油改性的聚乙烯醇基水凝胶,其中,x代表pva相对于水的质量百分比,如x=2%、4%、6%、8%、10%,y代表甘油相对于水的体积百分比,如0%、1%、2%、3%、4%;47.pc-glx指定聚乙烯醇/壳聚糖水凝胶,其中,x代表甘油相对于水的体积百分比,如x=0%、1%、2%、3%、4%、5%。48.实施例1制备水凝胶pvax-gly49.以pva6-gl3为例,其制备方法包括以下步骤:50.(1)称取pva12.0g、甘油6ml、去离子水200ml置于耐高压瓶中,摇匀,在120℃高温高压下,加热20min,转移至磁力搅拌器中80℃搅拌直至颗粒完全溶解,随后超声去泡,用吸管除去杂质,得到混合溶液;51.(2)将混合溶液倒入塑料正方形(24cm×24cm)平板模具中,室温条件下冷却48-72h成型;52.(3)将制备好的薄膜放入真空干燥箱中干燥,得到干燥薄膜;53.(4)干燥完成取出冷却后放入水中,每隔24h换水,浸泡72h后,得到水凝胶pva6-gl3。54.实施例2制备水凝胶pc-glx55.本发明的水凝胶pc-gl2的制备方法,包括以下步骤:56.(1)称取壳聚糖0.25g、聚乙烯醇12.3g、甘油2ml、醋酸0.925ml、去离子水200ml置于耐压瓶中120℃加热20min,随后转移至磁力搅拌器中80℃搅拌1h至颗粒完全溶解,超声去泡,得到混合溶液;57.(2)将混合溶液倒入塑料正方形平板模具中室温条件下冷却72h成型;58.(3)将制备好的薄膜放入真空干燥箱中(120℃、真空度0.1atm)12h得到干燥薄膜;59.(4)真空干燥完成取出冷却后放入水中,每隔24h换水,浸泡72h后,得到水凝胶。60.实施例3水凝胶的性能考察61.3.1力学性能测试62.采用电动冲片机将水凝胶薄膜裁出规定形状,使用千分尺测量水凝胶薄膜的厚度(精度≤1um),对每个测试样品的不同部位进行5次重复测量,得到平均厚度值。参照《gb/t 1040.3-2006塑料拉伸性能的测定》,使用微机控制电子万能试验机来测量水凝胶薄膜的机械性能(拉伸强度ts,断裂伸长率e,杨氏模量ym),实验中应变速率均保持为500mm/min。每个样品一式三份进行重复测量,取三组平均值得到最终的结果。63.3.1.1水凝胶pvax-gly64.(1)处理温度影响65.按照实施例1的方法,制备水凝胶pva6-gl3,区别在于处理步骤(3)中的处理温度分别选择60℃、80℃、100℃、120℃、140℃,结果如下:66.表1不同处理温度下pva6-gl3水凝胶薄膜的力学性能[0067][0068]注:同一列表中标记的不同字母显示出显著差异(n=3,p<0.05)[0069]由上表可知,随着处理温度的增加pva6-gl3水凝胶的最大力、拉伸强度以及最大力时的能量呈现出先上升后下降的趋势。在120℃时出现峰值,拉伸强度可达26.31±0.27mpa,最大力时的能量为4859.72±753.92n×mm。温度的升高可能加速薄膜的失水,促进pva分子间氢键的形成,甘油的存在能有效调节pva分子间形成的晶区的大小,形态与数量。在120℃时,可能是在甘油存在条件下,pva分子链间的网络结构最有利于力学性质的。但随着温度的继续升高达到140℃时,pva6-gl3水凝胶的机械性能反而下降,断裂拉伸应变下降至265.96±26.84%,这是由于pva6-gl3水凝胶长时间处于高温环境下,结构中的甘油也会蒸发,随着甘油的减少,pva分子间的结构网络会改变,力学性能发生改变。[0070](2)处理时间影响[0071]按照实施例1的方法,制备水凝胶pva6-gl3,区别在于步骤(3)中的处理时间分别选择0.5h、1h、2h、4h、6h、8h、10h、12h。[0072]表2不同处理时间下pva6-gl3水凝胶薄膜的力学性能[0073][0074]注:同一列表中标记的不同字母显示出显著差异(n=3,p<0.05)[0075]由上表可知,pva6-gl3水凝胶的力学性能随着处理时间增加而增强,当干燥时间为12h时,凝胶的拉伸强度与最大力时的能量达到峰值,这是由于干燥时间的延长,更加高效除去水分,使水凝胶网络结构中的微晶有充分时间形成,氢键结合增高,分子间作用力增强,使凝胶的网络结构越来越致密。[0076](3)pva加入量影响[0077]按照实施例1的方法,制备水凝胶pvax-gl3,pva的加入量分别选择相对于水的质量百分比为2%、4%、6%、8%、10%。[0078]表3 pvax-gl3水凝胶薄膜的力学性能[0079][0080]注:同一列表中标记的不同字母显示出显著差异(n=3,p<0.05)[0081]上表显示在120℃下干燥1h的pvax-gl3水凝胶膜在不同pva加入量下的拉伸强度、断裂拉伸应变、拉伸弹性模量以及最大力时的能量。pva6-gl3水凝胶拉伸强度表现出最大值,可达19.82±0.90mpa,在同一条件下,pva含量的增加,使分子间与分子内的氢键增多,分子间的作用力增强,提高了pvax-gl3凝胶的力学性能。但随着pva含量的继续增加,pvax-gl3的力学性能呈现出下降的趋势,这是由于体系中pva溶液的浓度增加,pva分子链增多,分子间作用力增强,运动阻力增大,使得pvax-gl3溶液粘度增加,晶区形成受到阻碍,使得凝胶的三维网络结构不稳定。另外,随着pva量的增加,甘油量随之减少,甘油的减少可能会影响pva分子间网络中形成的晶区的大小,形状以及数量,进而影响水凝胶的力学性质。[0082](4)甘油加入量影响[0083]按照实施例1的方法,制备水凝胶pva6-gly,甘油的加入量分别选择体积比为0%、1%、2%、3%、4%。[0084]表4 pva6-gly水凝胶薄膜的力学性能[0085][0086]注:同一列表中标记的不同字母显示出显著差异(n=3,p<0.05)[0087]上表显示pva6-gly水凝胶膜在不同甘油添加量下,120℃高温干燥12h所呈现出的拉伸强度、断裂拉伸应变、拉伸弹性模量以及最大力时的能量。随着甘油含量的增加,pva6-gly水凝胶膜的拉伸强度以及最大力时的能量呈现出先上升后下降的趋势,pva6-gl3水凝胶拉伸强度和最大力时的能量呈现最大值。与传统水凝胶(拉伸强度为0.2-10mpa)相比,pva6-gly水凝胶更加坚韧,更加具有弹性,富有优异的机械性能,其中pva6-gl3的拉伸强度可达26.31±0.27mpa,最大力时的能量为4859.72±753.92n×mm。[0088]表5 pva6-glx水凝胶薄膜的溶胀性能[0089][0090]注:同一列表中标记的不同字母显示出显著差异(n=3,p<0.05)[0091]上表中数据可以看出,经过120℃高温干燥12h后,随着甘油含量的增加,pva6-gly水凝胶的溶胀率呈现明显的下降趋势。分析其原因,是由于甘油的添加使得pva凝胶的结构中形成细小的晶区,而晶区分布的极为密集,使得交联网络越致密,网格间空隙变小,大量的水分子无法扩散,因此出现溶胀率逐渐下降的趋势。[0092](5)扫描电子显微镜(sem)[0093]裁取25mm2(5mm×5mm)正方形大小的水凝胶置于液氮中30min,经冷冻干燥处理24h后,喷金1min,在氩气环境下,采用扫描电子显微镜(sem)观察样品表面微观结构。[0094]如图2所示,可以看出水凝胶表面光滑,没有气孔以及其他孔洞结构,甘油的添加有利于水凝胶形成稳定致密的三维网络结构,这也验证了甘油的添加可以显著提升力学性质。大部分已经报道的通过冻融方法制备的pva水凝胶表面具有大量的孔洞,这也是降低力学性能的一个原因。[0095]3.1.2pc-glx水凝胶的力学性能[0096](1)甘油加入量的影响[0097]按照实施例2的方法,制备水凝胶pc-glx,甘油的加入量分别选择体积比为0%、1%、2%、3%、4%、5%。[0098]表6 pc-glx水凝胶薄膜的力学性能[0099][0100]上表显示水凝胶膜在添加不同甘油含量下的拉伸强度、断裂拉伸应变以及拉伸弹性模量。随着甘油含量的增加,pc-glx水凝胶膜的拉伸强度呈现出先升高后下降的趋势,pc-gl3水凝胶拉伸强度呈现峰值。与文献中记载的一般水凝胶(拉伸强度为0.2-10mpa)相比,pc-gl3水凝胶表现出优异的机械性能,拉伸强度为27.99±1.40mpa。甘油很可能在高温真空干燥过程中起了某种物理或化学的作用,对pva和cs的网络结构、分子间的力学作用起到了影响。[0101]表7 pc-glx水凝胶薄膜的溶胀性能[0102][0103]注:同一列表中标记的不同字母显示出显著差异(n=3,p<0.05)[0104]高温120℃真空干燥制成的pc-glx水凝胶薄膜在经过晾干失水重新在水中浸泡24h平衡后,通过表6结果可知,随着甘油含量的增加pc-glx水凝胶的溶胀率呈现下降趋势。这是由于随着甘油添加量的增加使得pva和cs的交联网络越致密,网格间空隙变小,缔合的水分子减少,故呈现出溶胀率逐渐下降的趋势,薄膜的抗水性能会出现明显改善。[0105](2)循环拉力数据分析[0106]采用电动冲片机将水凝胶pc-gl2、pc-gl3、pc-gl4裁成均一形状,测量三个位置厚度并取平均值,利用拉力机设置拉伸率分别为50%、100%、200%、250%、300%进行五次循环拉伸。[0107]结果如图3所示,上述三种水凝胶薄膜的机械性能较好,但在伸长率为50%、100%、200%、250%、300%五次循环拉伸过程中,pc-gl3水凝胶薄膜在循环拉伸过程中均表现出了优于其他两组的力学性能,这也与上述力学性能测试中拉伸强度结果相一致。这一现象表明相对于去离子水甘油添加量为3%的体积百分比这一较优含量下,甘油增强了动态化学键作用。壳聚糖在水凝胶网络结构内部生成的共价键与甘油增强的分子网络结构之间的动态化学键的作用使得水凝胶呈现较好的力学强度。三种薄膜在循环拉伸过程中横坐标显示的应变数值均未回到原点,是由于在连续循环拉伸过程中,动态化学键的断裂较多且重新形成的过程较慢,使得水凝胶在循环拉伸过程中不能立即恢复形变。[0108](3)扫描电子显微镜(sem)[0109]取5mm×5mm大小水凝胶置于液氮中20min,冷冻干燥处理24h后,在样品表面喷金1min,氩气环境下,采用扫描电子显微镜(sem)观察样品表面微观结构,放大倍数为5000倍。[0110]pc-glx水凝胶薄膜经过冷冻干燥过程将水凝胶网络结构中的冰晶升华,观察其内部形貌结构、分子链交联结构。图4中可以看出凝胶膜sem上的不明显的孔状结构,再次说明甘油的添加有利于水凝胶内部致密结构的产生。这表明以pva作为一级结构网络,壳聚糖作为二级结构网络,网络与网络间交错排列,以及随着甘油的添加使得分子网络交联作用增强伴随着壳聚糖形成的酰胺共价键作用下,最终使得水凝胶的分子网络呈现致密结构,也导致了上述溶胀率的降低。甘油在膜中一般是起减弱分子间的相互作用力的,但是在高温真空下到底对pva链的结晶起到一定的作用。[0111]以上所述,仅为本发明的具体实施方式,但本发明的保护范围并不局限于此,任何熟悉本技术领域的技术人员在本发明揭露的技术范围内,可轻易想到变化或替换,都应涵盖在本发明的保护范围之内。因此,本发明的保护范围应以所述权利要求的保护范围为准。 |
【本文地址】