| R语言用隐马尔可夫Profile HMM模型进行生物序列分析和模拟可视化 | 您所在的位置:网站首页 › 模拟预测模型怎么用 › R语言用隐马尔可夫Profile HMM模型进行生物序列分析和模拟可视化 |
R语言用隐马尔可夫Profile HMM模型进行生物序列分析和模拟可视化
|
对于一系列观察到的掷骰,我们可以使用维特比算法建立最可能的隐藏状态序列(包括最有可能发生骰子切换的时间)。在 Durbin 等人 (1998) 第 3.2 章给出的示例中,观察到的 300 次滚动的序列为:
一些可观察到的 6 簇表明加载的骰子在某个阶段出现了,但是骰子切换是什么时候发生的呢?在下面的代码中,Viterbi 算法用于在给定模型的情况下找到最可能的隐藏状态序列。 将预测路径与实际隐藏序列进行比较,Viterbi 算法并不遥远:
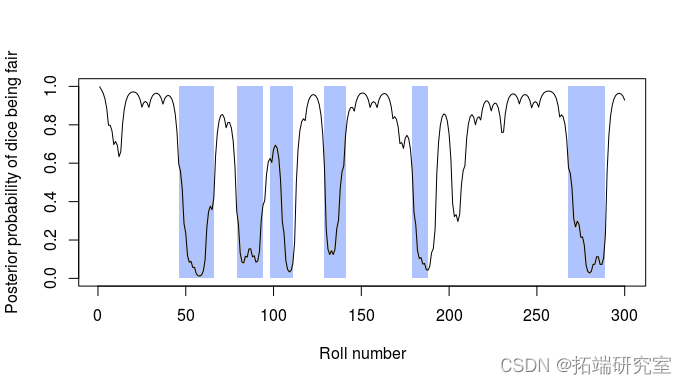
我们还可以使用 forward 和/或 backward 算法计算给定模型的序列的完整概率和后验概率: 图 2:300 次掷骰子的后验状态概率。 这条线显示了骰子在每次掷骰时都是公平的后验概率,而灰色矩形显示了使用加载骰子的实际周期。有关详细信息,请参见 Durbin 等人 (1998) 第 3.2 章。
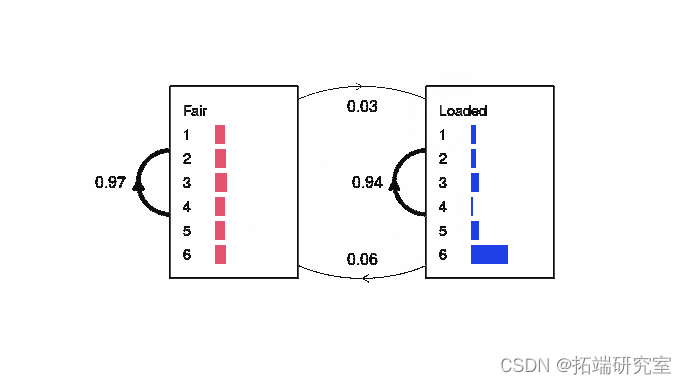
从一组训练序列构建HMM 。以下代码从我们的单个掷骰子序列及其已知状态路径(存储为序列的“名称”属性)中派生出一个简单的 HMM。
图 3:从 300 次掷骰子序列派生的简单 HMM。 如图 所示,转换概率显示为加权线,发射概率显示为水平灰色条。 尽管训练数据仅由一个序列组成,但这似乎与实际模型相当接近。人们通常会从许多此类序列的列表中导出 HMM(因此输入参数是列表而不是向量的原因),但为清楚起见,此示例已简化。 HMM隐马尔可夫模型Profile HMM马尔可夫模型是标准 HMM 的扩展,其中转移概率是 特定于位置的。也就是说,它们可以在序列中的每个点发生变化。这些模型通常比其简单的 HMM 模型具有更多的参数,但对于序列分析可能非常强大。Profile HMM 的前身通常是多序列比对。 图 4 将上面列出的三种状态类型分别显示为圆形、菱形和矩形。这些状态由图中的加权线所示的转移概率链接。 考虑来自 Durbin 等人 (1998) 第 5.3 章的氨基酸序列的小部分比对:
位置特定模式包括在位置 1 观察到“V”和在位置 3 观察到“A”或“G”的高概率。 以下代码从 globin 数据派生出 HMM 轮廓并绘制模型: glsosdbgnss.PsdHMMf |



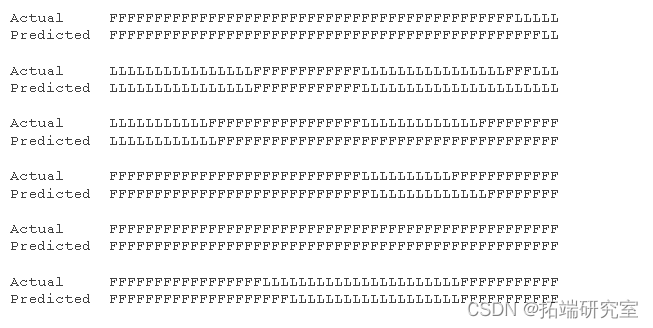




【本文地址】