| 根据文件名前缀及基因名称批量提取基因序列 | 您所在的位置:网站首页 › 文件夹批量加前缀代码怎么加 › 根据文件名前缀及基因名称批量提取基因序列 |
根据文件名前缀及基因名称批量提取基因序列
|
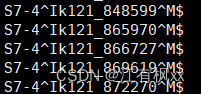
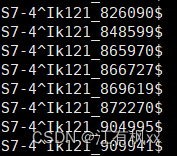
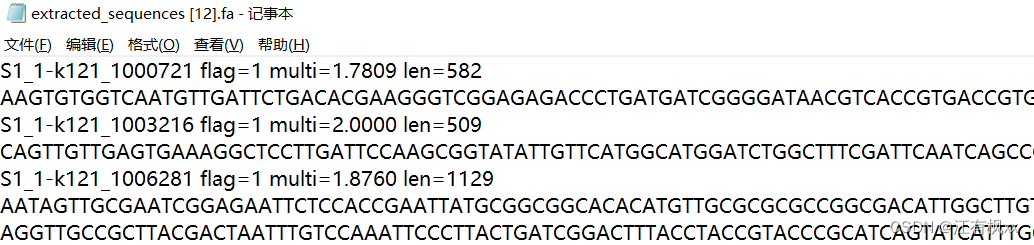
我一整个大愚蠢,这种东西非常非常基础的东西就应该用别人已经成熟的工具解决(最好是用seqtk啦) 比如: (6条消息) 如何根据 ID 快速从 fastq 文件中提取序列_根据id提取fastq序列_klcola的博客-CSDN博客 (6条消息) 根据基因名称批量提取基因序列_wt12138的博客-CSDN博客 但我居然不先查资料,愚蠢地造轮子(怒!)。算是学习了,而且对于多文件的操作或许会更快吧(自我安慰道.jpg)。。。 Step1. 序列文件的预处理(如果需要的话,因为我是经过了excel操作后导出的,可能因此导致了这愚蠢的问题) # window操作后,有时换行符会和linux里的不一样,这时便需要手动更改 sed -i 's/\r$//' seq_info.txt # 换行符 sed 's/\^/ /' seq_info.txt # 分列符 cat -A seq_info.txt # 检查换行符对不对如果经cat -A检验是这样,就不行 经cat -A检验形如这样,就行 以下这个代码实现的是: seq_info.txt是一个两列无表头的文件,第一列放文件名前缀,第二列放序列名。这是之前根据某条件比对筛选后,合并各文件比对结果的文件(之所以合并,是为了其他分析的快捷性)。 result/contigs是contig文件的路径,有很多.fa文件 根据seq_info.txt文件提取文件夹中的序列,输出到extracted_seqences.fa while read prefix sequence_name; do grep -A 1 "\b$sequence_name\b" result/contigs/"$prefix".fa \ | sed "s/^>\(.*\)$/$prefix-\1/" done < seq_info.txt > extracted_sequences.fa得到的结果文件形如:
|
【本文地址】
公司简介
联系我们