| 简化基因组数据分析实战 | 您所在的位置:网站首页 › 基因组测序数据分析怎么写 › 简化基因组数据分析实战 |
简化基因组数据分析实战
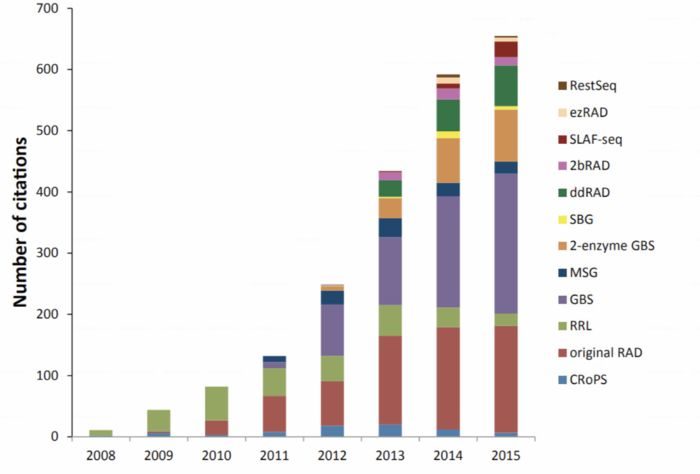
 RAD-seq和GBS是什么关系 简化基因组的测序方法RAD-Seq(restriction site-associated DNA sequencing)最开始指的是2008年发表在PLOS ONE上“Rapid SNP discovery and genetic mapping using sequenced RAD markers”提出的方法,目前该文章的引用已经达到1200 ,现在指代的是一系列基于限制性内切酶的测序技术。同样在概念上被引申的还有GBS(genotyping-by-sequencing),只不过GBS的名字不能让你直接把它和限制性内切酶联想起来.总之,如果现在公司给你推荐GBS或RAD-seq时,可能未必和你想的一样,你需要仔细问下他们的建库手段。毕竟手段不同,你的实验设计,操作和结果都会发生变化。这是RAD-seq相关方法的历年引用情况
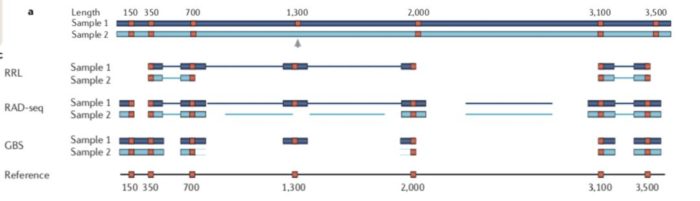
RAD-seq虽说方法很多,但是文库构建流程大致如下,不同方法在其中某些步骤存在差异 起始基因组DNA量:能否允许降解FNA限制性内切酶酶解:限制酶种类,数量酶切位点结合接头:接头类型酶解片段大小选择:直接选择,间接选择添加barcode混池:视v接头而异测序类型选择:单端,双端两者的差异在于,1)是现进行酶切然后随机破碎,最后仅选择存在酶切位点片段测序;2)也是酶切,但是后续直接选择合适大小的片段测序。 因此相对于1)测序的位点平均会少一点,也就会导致同一批样本后者利用率低于前者。无参考基因组更推荐前者,而不是后者。
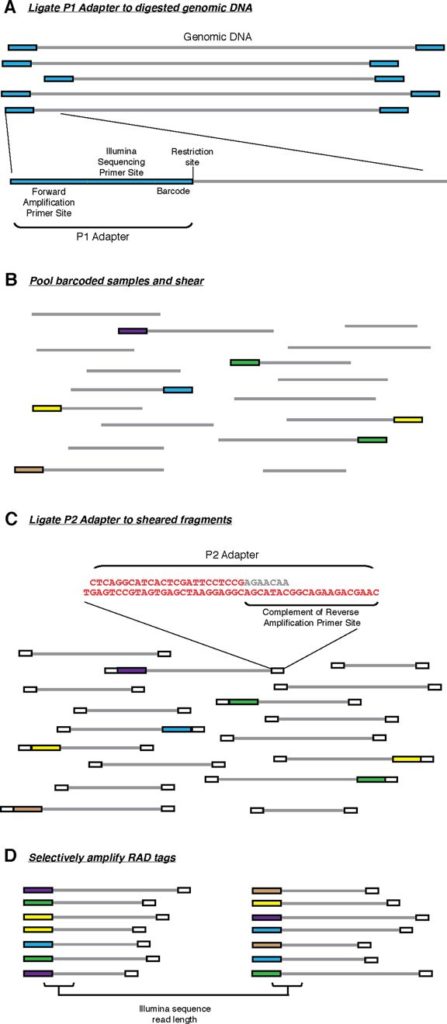
最先提出的RAD-seq技术流程,也就是RAD-seq的冠名技术,分为如下几步: 基因组DNA用限制性内切酶裂解, 然后连接到P1接头。P1接头里含有正向扩增和Illumina测序引物位点,以及4~5 bp 的核酸barcode. barcode至少大于3 bp。之后接头连接的片段(adapter-ligated fragments)混池,随机打断DNA随后连接到P2接头,反向扩增扩展引物无法连接P2. P2是一种Y型接头,包含P2反向扩增引物位点的反向互补序列,使得不含P1接头的片段无法扩增。(Y型接头的工作原理)最后仅有同时含P1和P2接头的片段能够上机测序。
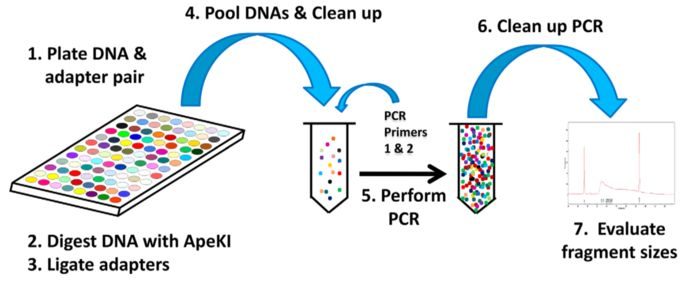
GBS比原始的RAD-seq步骤更加简单 将不同样本和含不同barcode接头成对放在平板里使用ApeKI限制酶进行酶解使用T4连接酶,将接头连接到片段两端因酶切产生的粘末端(stcky end)将含不同barcode的样本混池,随后过片段长度筛选柱,过滤尚未反应的接头加入PCR引物,进行PCR扩增这里没有直接对片段进行筛选,但是PCR扩增时优先扩增小片段
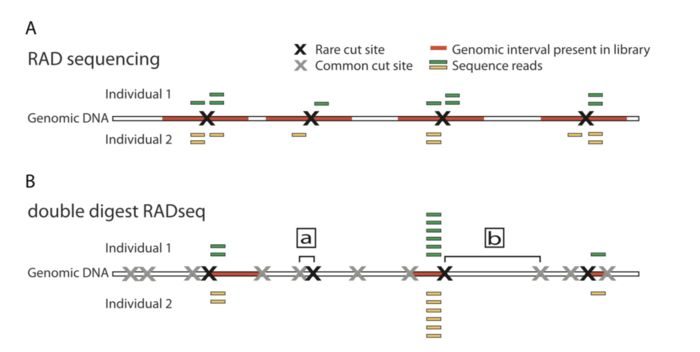
Genotyping-by-Sequencing流程 ddRAD-seqddRAD-seq和GBS相似,两者都不需要在加接头后进行随机打碎,GBS通过PCR扩增的方式过滤了大片段,而ddRAD-seq通过双酶切的方式,然后筛选固定长度来选择合适大小的片段
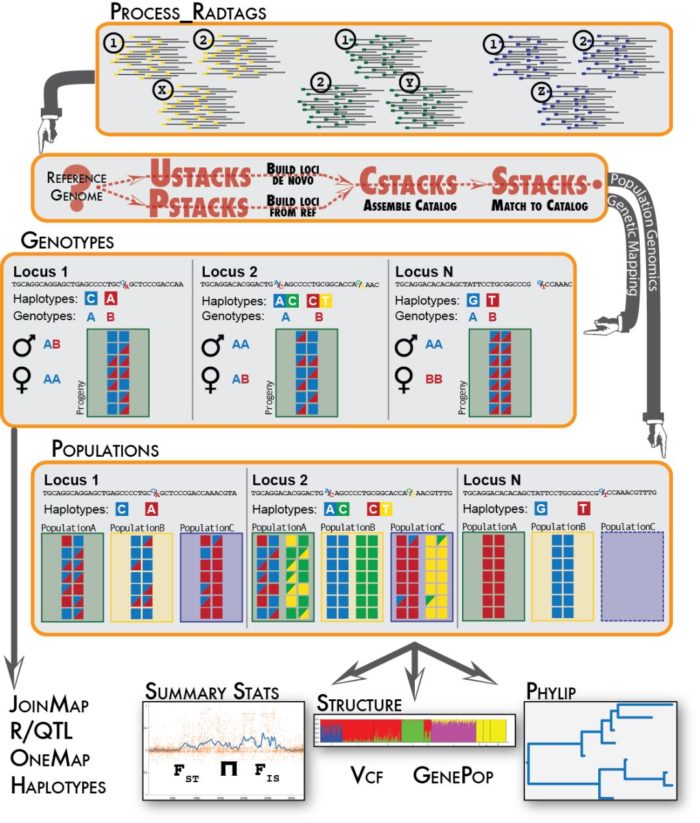
ddRAD-seq和RAD-seq的不同 常见方法的比较 其实这些RAD-seq文库制备方法可以简单的分为两类: 1)对单酶切位点邻近片段测序,如最初的RAD-seq2)对酶切位点两翼片段测序,如Genoytping-by-Sequencing下面是常见的物RAD-seq方法比较 方法原始RAD2bRADGBSddRADezRAD控制位点的方法选择限制酶选择限制酶选择限制酶选择限制酶和片段大小选择阈值选择限制酶和片段大小选择阈值位点数/Mb30~50050~10005~400.3~20010~800位点长度300bp 或1kb contig33–36 bp< 300 bp< 300 bp 01-clean-data/process_radtags.lane1.oe &解释下参数,虽然大部分已经很明了: -p为原始数据存放文件夹 -b为barcode和样本对应关系的文件 -o为输出文件夹 -e为建库所用的限制性内切酶 --inline_null表示barcode的位置在单端的read中 -c表示数据清洗时去除表示为N的碱基 -q表示数据清理时要去除低质量碱基 -r表示要抢救下barcode和RAD-tag。
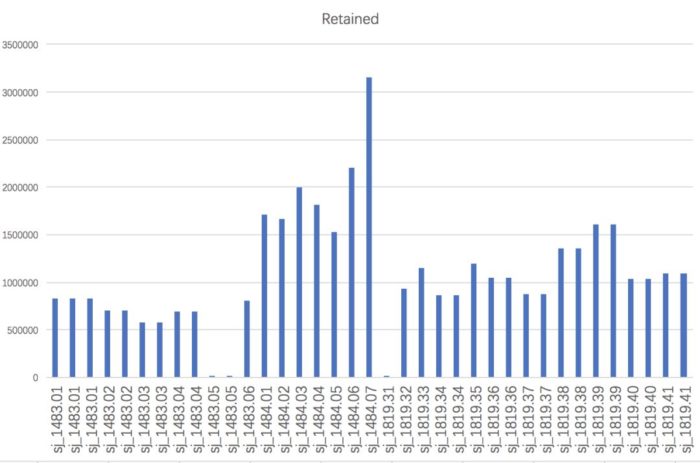
这一步需要留意自己的单端测序,还是双端测序,barcode是在read中,还是在FASTQ的header中,是否还需要去接头序列,是否是双酶切建库等。 另外这一步比较耗时,尽量脱机运行或者提交到计算节点中,不然突然断网导致运行终止就更浪费时间了。 将运行结果记录到日志文件中,方便后期检查报错。 运行结束后,在01-clean-data下会有除了process_radtags.lane1.oe外,还会有process_radtags.lane1.log,前者记录每条lane的数据保留情况,后者记录每个样本的数据保留情况。可以将后者复制到Excel表格中,用柱状图等方法直观了解
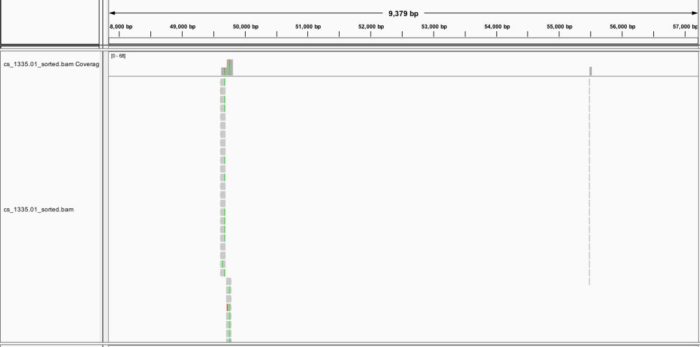
从图中可以发现,”sj_1483.05”和”sj_1819.31”几乎没有read留下来,这能是建库上导致的问题,我们需要将其从“info/popmap.tsv”中删掉或者用“#”注释掉(推荐后者)。 在数据预处理这一步,stacks还提供process_shortreads,clone_filter, kmer_filter用于处理cDNA文库和随机切割的DNA文库,如果是RAD-seq不需要用到。 如果是双端测序,stacks1.47只能通过cat合并两个数据,而不能有效的利用双端测序提供的fragment信息。stacks似乎可以,我之后尝试。 后续就要区分为有参和无参两条路了。 参考资料 Deriving genotypes from RAD-seq short-read data using StacksGenome-wide genetic marker discovery and genotyping using next-generation sequencing第二步:序列比对这一步之后,分析流程就要根据是否有参考基因组分别进行分析。无参考基因组需要先有一步的 de novo 组装,产生能用于比对的contig。有参考基因组则需要考虑基因组的质量,如果质量太差,则需要进一步以无参分析作为补充。 参考基因组主要用于区分出假阳性的SNP,将snp与附近其他共线性的snp比较来找出离异值,这些离异值大多是因为建库过程所引入的误差,如PCR的链偏好性扩增。 无论是何者,我们一开始都只能用其中的部分数据进行参数测试,根据不同的参数结果作为反馈,进行调优,这一步根据你的运气和经验,还有你的算力,时间不定。毕竟超算一天,普算一年。 有参考基因组三刺鱼是可从ensemblgenomic上搜索到到参考基因组信息 http://asia.ensembl.org/Gasterosteus_aculeatus/Info/Index但是质量非常一般,仅仅是contig程度,只能说是凑合使用了。 建立索引数据库stacks不直接参与比对,而是处理不同比对软件得到的BAM文件,因此你可以根据自己的喜好选择比较工具。目前,基因组比对工具都是首选BWA-mem,所以这里建立bwa的索引 # 位于项目根目录下 cd reference/genome wget -q ftp://ftp.ensembl.org/pub/release-91/fasta/gasterosteus_aculeatus/dna/Gasterosteus_aculeatus.BROADS1.dna.toplevel.fa.gz gzip -d Gasterosteus_aculeatus.BROADS1.dna.toplevel.fa.gzcd .. mkdir -p index/bwa/ genome_fa=genome/Gasterosteus_aculeatus.BROADS1.dna.toplevel.fa bwa index -p index/bwa/gac $genome_fa &> index/bwa/bwa_index.oe # 结果如下 |-- genome | |-- Gasterosteus_aculeatus.BROADS1.dna.toplevel.fa |-- index |-- bwa |-- bwa_index.oe |-- gac.amb |-- gac.ann |-- gac.bwt |-- gac.pac |-- gac.sa小样本参数调优这一步是为了调整比对工具处理序列相似性的参数,保证有绝大多数的read都能回帖到参考基因组上,因此参数不能太严格,能容忍遗传变异和测序误差,也不能太宽松,要区分旁系同源位点。对于BWA-MEM而言,几个和打分相关的参数值得注意: -B: 不匹配的惩罚, 影响错配数,默认是4-O: 缺失和插入的gap打开的惩罚,影响InDel的数目,默认是[6,6]-E: gap延伸的惩罚,长度k的gap惩罚为’{-O} {-E}*k’, 默认是[1,1]-L: soft clip的惩罚,也就是read两端直接切掉碱基来保证匹配,默认是[5,5]对于参考基因组质量比较高,且研究物种和参考基因组比较近,那么参数调整没有太大必要性。如果质量不要,或者所研究物种和参考基因组有点距离,那么就需要注意不同参数对结果的影响,必要时使用IGV人工检查。 让我们先以默认参数开始,处理其中一个样本 # 位于项目根目录下# 在测试文件下按照参数创建文件夹 mkdir -p test/ref/{alignment-bwa,stacks-bwa} ## bwa-mem比对 sample=cs_1335.01fq_file=01-clean-data/$sample.fq.gz bam_file=test/ref/alignment-bwa/${sample}_default.bam bwa_index=reference/index/bwa/gac bwa mem -M $bwa_index $fq_file | samtools view -b > $bam_file &这里的bwa mem使用了-M参数,通常的解释这个参数是为了和后续的Picard标记重复和GATK找变异兼容。 进一步的解释,不用-M,split read会被标记为SUPPLEMENTARY, 使用该选项则是标记为SECONDARY(次要),即不是PRIMARY(主要),既然不是主要比对,所以就被一些工具忽略掉。如果是SUPPLEMENTARY就有可能在标记重复时被考虑进去。其中split read是嵌合比对的一部分,具体概念见SAM格式解释。 对于比对结果,可以用samtools stats和samtools flagstat查看一下质量 samtools flagstat test/ref/alignment-bwa-default/cs_1335.01_default.bam #1310139 0 in total (QC-passed reads QC-failed reads) #7972 0 secondary #0 0 supplementary #0 0 duplicates#1271894 0 mapped (97.08% : N/A)97.08%的比对率应该是很不错了,不过可以尝试降低下错配和gap的惩罚,看看效果 # 位于项目根目录下 sample=cs_1335.01fq_file=01-clean-data/$sample.fq.gz bam_file=test/ref/alignment-bwa/${sample}_B3_O55.bam bwa_index=reference/index/bwa/gac bwa mem -M -B 3 -O 5,5 $bwa_index $fq_file | samtools view -b > $bam_file & samtools flagstat test/ref/alignment-bwa-default/cs_1335.01_default.bam #1309830 0 in total (QC-passed reads QC-failed reads) #7663 0 secondary #0 0 supplementary #0 0 duplicates#1272297 0 mapped (97.13% : N/A)也就提高了0.05%,所以就用默认参数好了。通过IGV可视化,可以了解简化基因组的read分布是比较稀疏,10k中可能就只有2个。
得到的BAM文件使用pstack中找snp, # 位于项目根目录下 sample=cs_1335.01 sample_index=1 bam_file=test/ref/alignment-bwa/${sample}_B3_O55.bam log_file=test/ref/stacks-bwa/$sample.pstacks.oe pstacks -t bam -f $bam_file -i $sample_index -o test/ref/stacks-bwa/ &> $log_file这里的参数也很简单,-t用来确定输入文件的格式,-f是输入文件,-i对样本编序,-o指定输出文件夹。除了以上几个参数外,还可用-p指定线程数,-m来制定最低的覆盖度,默认是3.还可以用--model_type [type]制定模型。 最后得到$sample.tags.tsv.gz, $sample.models.tsv.gz, $sample.snps.tsv.gz, 和 $sample.alleles.tsv.gz共4个文件,以及一个日志文件。参数评估的主要看日志文件里的几个指标: 实际使用的alignment数因soft-clipping剔除的alignment数,过高的话要对比对参数进行调整每个位点的平均覆盖度,过低会影响snp的准确性。这里仅仅用了一个样本做测试,实际上要用10个以上样本做测试,看平均表现, 全数据集处理在使用小样本调试完参数,这部分参数就可以应用所有的样本。除了比对和使用pstacks外,还需要用到 cstacks根据位置信息进一步合并成包含所有位点信息的目录文件,之后用 sstacks从 cstacks创建的目录文件搜索每个样本的位点信息。代码为 cstacks -p 10 --aligned -P 03-alignment-stacks/ -M info/popmap.tsv # 以其中一个样本为例 sample=cs_1335.01 log_file=$sample.sstacks.oe sstacks --aligned -c 03-alignment-stacks/batch_1 -s 03-alignment-stacks/$sample -o 03-alignment-stacks/ &> 03-alignment-stacks/$log_file你可以写一个shell脚本处理,不过我现在偏好用snakemake写流程,如下: INDEX = "reference/index/bwa/gac" SAMPLES, = glob_wildcards("01-raw-data/{sample}.fq.gz") INDEX_DICT = {value: key for key, value in dict(enumerate(SAMPLES, start=1))} FQ_FILES = expand("01-clean-data/{sample}.fq.gz", sample=SAMPLES) BAM_FILES = expand("02-read-alignment/ref-based/{sample}.bam", sample=SAMPLES) SNP_TSVS = expand("03-alignment-stacks/{sample}.snp.tsv.gz", sample=SAMPLES) CATALOG = "03-alignment-stacks/batch_1.catalog.snps.tsv.gz" MATCHES = expand("03-alignment-stacks/{sample}.matches.tsv.gz", sample=SAMPLES) rule all: input: BAM_FILES, rule bwa_mem: input: "01-clean-data/{sample}.fq.gz" params: index = INDEX, mismatch = "3", gap = "5,5" threads: 8 output: "02-read-alignment/ref-based/{sample}.bam" shell:""" mkdir -p 02-read-alignment bwa mem -t {threads} -M -B {params.mismatch} -O {params.gap} {params.index} {input} \ | samtools view -b > {output} """ rule pstacks: input: "02-read-alignment/ref-based/{sample}.bam" params: index = lambda wildcards: INDEX_DICT.get(wildcards.sample), outdir = "03-alignment-stacks/" threads: 8 output: "03-alignment-stacks/{sample}.snps.tsv.gz", "03-alignment-stacks/{sample}.tags.tsv.gz", "03-alignment-stacks/{sample}.models.tsv.gz", "03-alignment-stacks/{sample}.alleles.tsv.gz" log: "03-alignment-stacks/{sample}.pstacks.oe" shell:""" mkdir -p 03-alignment-stacks pstacks -p {threads} -t bam -f {input} -i {params.index} -o {params.outdir} &> {log} """ # A catalog can be built from any set of samples processed by the ustacks or pstacks programs rule cstacks: input: "info/popmap.tsv",expand("03-alignment-stacks/{sample}.snps.tsv.gz", sample=SAMPLES) threads: 10 output: "03-alignment-stacks/batch_1.catalog.alleles.tsv.gz", "03-alignment-stacks/batch_1.catalog.snps.tsv.gz", "03-alignment-stacks/batch_1.catalog.tags.tsv.gz" shell: "cstacks -p {threads} --aligned -P 03-alignment-stacks/ -M {input[0]}" # Sets of stacks, i.e. putative loci, constructed by the ustacks or pstacks programs # can be searched against a catalog produced by cstacks. rule sstacks: input: "03-alignment-stacks/{sample}.snps.tsv.gz", "03-alignment-stacks/{sample}.tags.tsv.gz", "03-alignment-stacks/{sample}.models.tsv.gz", "03-alignment-stacks/{sample}.alleles.tsv.gz" params: catalog = "03-alignment-stacks/batch_1", sample = lambda wildcards: "03-alignment-stacks/" wildcards.sample output: "03-alignment-stacks/{sample}.matches.tsv.gz" log: "03-alignment-stacks/{sample}.sstacks.oe" shell:""" sstacks --aligned -c {params.catalog} -s {params.sample} -o 03-alignment-stacks &> {log} """将以上代码保存为Snakemake,没有集群服务器,就直接用 snakemake运行吧。因为我能在集群服务器上提交任务,所以用如下代码: snakemake --cluster "qsub -V -cwd" -j 20 --local-cores 10 &无参考基因组分析
基于参考基因组的分析不能体现出RAD-seq的优势,RAD-seq的优势体现在没有参考基因组时他能够提供大量可靠的分子标记,从而构建出遗传图谱,既可以用于基因定位,也可以辅助组装。 和全基因组随机文库不同,RAD-seq是用限制性内切酶对基因组的特定位置进行切割。这样的优点在于降低了 de novo 组装的压力,原本是根据overlap(重叠)来延伸150bp左右短读序列,形成较大的contig,而现在只是将相似性的序列堆叠(stack)起来。这样会产生两种分子标记:1)由于变异导致的酶切位点出现有或无的差异;2)同一个酶切位点150bp附近存在snp。 参数调优 这一步使用的核心工具是 ustacks和 cstacks,前者根据序列相似性找出变异,后者将变异汇总,同样需要使用小样本调整三个参数 -M, -n, -m。 ustacks的 M 控制两个不同样本等位基因(allele)之间的错配数,m 控制最少需要几个相同的碱基来形成一个堆叠(stack).最后一个比较复杂的参数是能否允许gap(--gap)。而 cstacks的 n 和 ustacks的 M等价。 因此,我们可以尝试在保证 m=3的前提,让 M=n从1到9递增,直到找到能让80%的样本都有多态性RAD位点,简称r80. Stacks提供了 denovo_map.pl来完成这部分工作,下面开始代码部分。 首先是从 info/popmap.tsv中挑选10多个样本用于参数调试。 cat popmap_sample.tsv cs_1335.01 cs cs_1335.02 cs cs_1335.19 cs pcr_1193.00 pcr pcr_1193.01 pcr pcr_1193.02 pcr pcr_1213.02 pcr sj_1483.01 sj sj_1483.02 sj sj_1483.03 sj wc_1218.04 wc wc_1218.05 wc wc_1218.06 wc wc_1219.01 wc然后为每个参数都准备一个文件夹 mkdir -p test/de-novo/stacks_M{1..9}然后先 测试 第一个样本 # 项目根目录 M=1 popmap=info/popmap_sample.tsv reads_dir=01-clean-data out_dir=test/de-novo/stacks_M${M} log_file=$out_dir/denovo_map.oe threads=8 denovo_map.pl -T ${threads} --samples ${reads_dir} -O ${popmap} -o ${out_dir} -M $M -n $M -m 3 -b 1 -S &> ${log_file} &代码运行需要一段比较长的时间,这个时候可以学习一下参数: -T 表示线程数, --samples表示样本数据所在文件夹,-O提供需要分析的样本名, -o是输出文件夹,之后就是-M, -n, -m这三个需要调整的参数, -b表示批处理的标识符, -S关闭SQL数据库输出。同时还有遗传图谱和群体分析相关参数,-p表示为亲本,-r表示为后代,-s表明群体各个样本。 为了确保下一步能顺利进行,还需要对oe结尾的日志文件的信息进行检查,确保没有出错 grep -iE "(err|e:|warn|w:|fail|abort)" test/de-novo/stacks_M1/denovo_map.oe以及以log结尾的日志中每个样本的平均覆盖度,低于10x的覆盖度无法保证snp的信息的准确性 之后对每个参数得到的原始数据,要用 populations过滤出80%以上都有的snp位点,即r80位点 # 项目根目录 for M in `seq 1 9` do popmap=info/popmap_sample.tsv stacks_dir=test/de-novo/stacks_M${M} out_dir=$stacks_dir/populations_r80 mkdir -p ${out_dir} log_file=$out_dir/populations.oe populations -P $stacks_dir -O $out_dir -r 0.80 &> $log_file & done确定参数 经过漫长的等待后,所有参数的结果都已经保存到特定文件下,最后就需要从之确定合适的参数,有两个指标: 多态性位点总数和r80位点数短读堆叠区的snp平均数尽管这些信息都已经保存在了相应的文本 test/de-novo/stacks_M[1-9]/populations_r80/batch_1.populations.log中,但是通过文字信息无法直观的了解总体情况,因此更好的方法是用R处理输出绘图结果。 第一步:提取每个参数输出文件中的log文件中SNPs-per-locus distribution(每个位点座位SNP分布图)信息.新建一个shell脚本,命名为,log_extractor.sh, 添加如下内容 #!/bin/bash # Extract the SNPs-per-locus distributions (they are reported in the log of populations). # ---------- echo "Tallying the numbers..." full_table=n_snps_per_locus.tsv header=\'#par_set\tM\tn\tm\tn_snps\tn_loci\' for M in 1 2 3 4 5 6 7 8 9 ;do n=$M m=3 # Extract the numbers for this parameter combination. log_file=test/de-novo/stacks_M${M}/populations_r80/batch_1.populations.log sed -n \'/^#n_snps\tn_loci/,/^[^0-9]/ p\' $log_file | grep -E \'^[0-9]\' > $log_file.snps_per_loc # Cat the content of this file, prefixing each line with information on this # parameter combination. line_prefix="M$M-n$n-m$m\t$M\t$n\t$m\t" cat $log_file.snps_per_loc | sed -r "s/^/$line_prefix/" done | sed "1i $header" > $full_table运行后得到 n_snps_per_locus.tsv用于后续作图。会用到两个R脚本 plot_n_loci.R和 plot_n_snps_per_locus.R,代码见最后,结果图如下 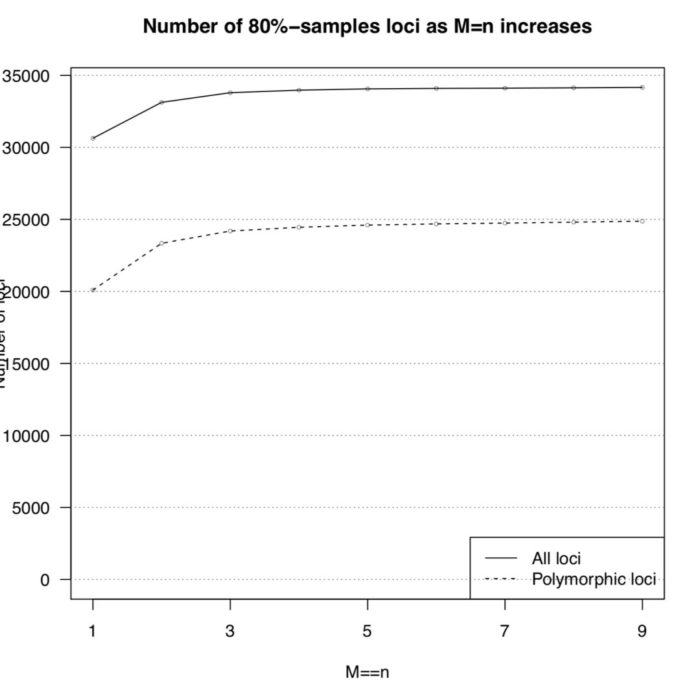 r80位点数量 r80位点数量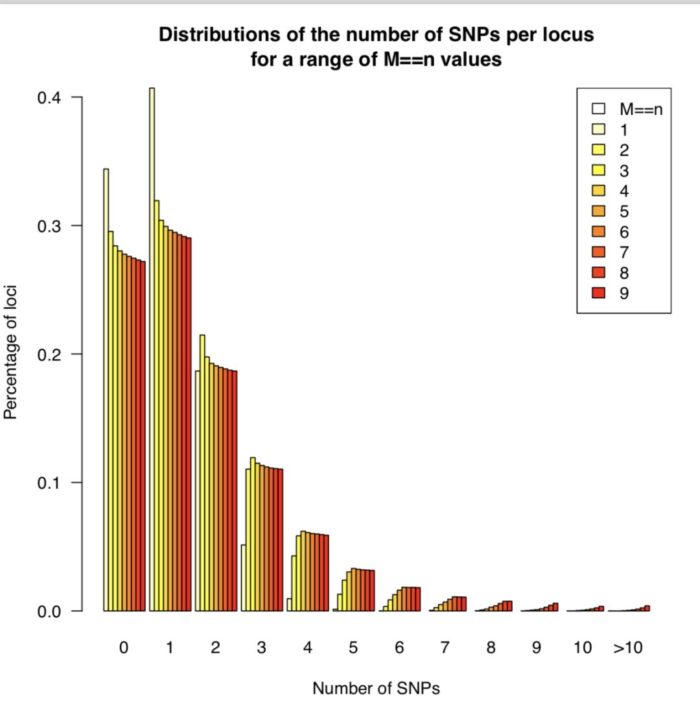 SNP分布 SNP分布从上图可以发现M=4以后,折线就趋于稳定,并且每个座位上的SNP分布趋于稳定,因此选择M=4作为全样本数据集的运行参数。 全数据集 de novo 分析和之前基于参考基因组分析的代码类型,只不过将序列比对这一块换成了 ustacks。尽管前面用来确定参数的脚本 denovo_map.pl也能用来批量处理,但它不适合用于大群体分析。 ustacks得到的结果仅能选择40~200个 覆盖度高 且 遗传多样性上有代表性的样本。 使用所有样本会带来计算上的压力,低频的等位基因位点也并非研究重点,并且会提高假阳性。综上,选择覆盖度比较高的40个样本就行了。 总体部分已经通过这三天讲完,明后天更新SNP过滤部分 代码Snakefile: SAMPLES, = glob_wildcards("01-clean-data/{sample}.fq.gz") INDEX_DICT = {value: key for key, value in dict(enumerate(SAMPLES, start=1)).items()} FQ_FILES = expand("01-clean-data/{sample}.fq.gz", sample=SAMPLES) # de novo MISMATCH = 4 # mismatch number of ustacks DE_NOVO_LOCI = expand("03-stacks-analysis/de-novo/{sample}.snps.tsv.gz", sample=SAMPLES) DE_NOVO_CATA = "03-stacks-analysis/de-novo/batch_1.catalog.snps.tsv.gz" DE_NOVO_MATS = expand("03-stacks-analysis/de-nono/{sample}.matches.tsv.gz", sample=SAMPLES) # ref-based INDEX = "reference/index/bwa/gac" BAM_FILES = expand("02-read-alignment/ref-based/{sample}.bam", sample=SAMPLES) REF_BASED_LOCI = expand("03-stacks-analysis/ref-based/{sample}.snps.tsv.gz", sample=SAMPLES) REF_BASED_CATA = "03-stacks-analysis/ref-based/batch_1.catalog.snps.tsv.gz" REF_BASED_MATS = expand("03-stacks-analysis/ref-based/{sample}.matches.tsv.gz", sample=SAMPLES) rule all: input: rules.de_novo.input, rules.ref_based.input rule de_novo: input: DE_NOVO_LOCI, DE_NOVO_CATA,DE_NOVO_MATS rule ref_based: input: REF_BASED_LOCI, REF_BASED_CATA, REF_BASED_MATS # de novo data analysis ## The unique stacks program will take as input a set of short-read sequences ## and align them into exactly-matching stacks (or putative alleles). rule ustacks: input: "01-clean-data/{sample}.fq.gz" threads: 8 params: mismatch = MISMATCH, outdir = "03-stacks-analysis/de-novo", index = lambda wildcards: INDEX_DICT.get(wildcards.sample) output: "03-stacks-analysis/de-novo/{sample}.snps.tsv.gz", "03-stacks-analysis/de-novo/{sample}.tags.tsv.gz", "03-stacks-analysis/de-novo/{sample}.models.tsv.gz", "03-stacks-analysis/de-novo/{sample}.alleles.tsv.gz", log: "03-stacks-analysis/de-novo/{sample}.ustacks.oe" shell:""" mkdir -p {params.outdir} ustacks -p {threads} -M {params.mismatch} -m 3 \ -f {input} -i {params.index} -o {params.outdir} &> {log} """ ## choose sample for catalog building rule choose_representative_samples: input: expand("03-stacks-analysis/de-novo/{sample}.ustacks.oe", sample=SAMPLES) output: "03-stacks-analysis/de-novo/per_sample_mean_coverage.tsv", "info/popmap.catalog.tsv" shell:""" for sample in {input};do name=${{sample##*/}} name=${{name%%.*}} sed -n "/Mean/p" ${{sample}} |\ sed "s/.*Mean: \(.*\); Std.*/\\1/g" |\ paste - - - |\ sed "s/.*/${{name}}\\t&" done | sort -k2,2nr > {output[0]} head -n 50 {output[0]} | tail -n 40 | cut -f 1 | sed \'s/\([0-9a-zA-Z]*\)_.*/&\t\1/\' > {output[1]} """ rule de_novo_cstacks: input: "info/popmap.catalog.tsv", expand("03-stacks-analysis/de-novo/{sample}.snps.tsv.gz", sample=SAMPLES) params: stacks_path = "03-stacks-analysis/de-novo/", mismatch = MISMATCH threads: 10 log:"03-stacks-analysis/de-novo/de_novo_cstacks.oe" output: "03-stacks-analysis/de-novo/batch_1.catalog.alleles.tsv.gz", "03-stacks-analysis/de-novo/batch_1.catalog.snps.tsv.gz", "03-stacks-analysis/de-novo/batch_1.catalog.tags.tsv.gz" shell:""" cstacks -p {threads} -P {params.stacks_path} -M {input[0]} \ -n {params.mismatch} &> {log} """ rule de-novo_sstacks: input: "03-stacks-analysis/de-novo/{sample}.snps.tsv.gz", "03-stacks-analysis/de-novo/{sample}.tags.tsv.gz", "03-stacks-analysis/de-novo/{sample}.models.tsv.gz", "03-stacks-analysis/de-novo/{sample}.alleles.tsv.gz" params: catalog = "03-stacks-analysis/de-novo/batch_1", sample = lambda wildcards: "03-stacks-analysis/de-novo/" wildcards.sample output: "03-stacks-analysis/de-novo/{sample}.matches.tsv.gz" log: "03-stacks-analysis/de-novo/{sample}.sstacks.oe" shell:""" sstacks -c {params.catalog} -s {params.sample} -o 03-stacks-analysis/de-novo &> {log} """ # reference-based data analysis ## read alignment with bwa-mem rule bwa_mem: input: "01-clean-data/{sample}.fq.gz" params: index = INDEX, mismatch = "3", gap = "5,5" threads: 8 output: "02-read-alignment/ref-based/{sample}.bam" shell:""" mkdir -p 02-read-alignment bwa mem -t {threads} -M -B {params.mismatch} -O {params.gap} {params.index} {input} \ | samtools view -b > {output} """ ## find variant loci rule pstacks: input: "02-read-alignment/ref-based/{sample}.bam" params: outdir = "03-stacks-analysis/ref-based/", index = lambda wildcards: INDEX_DICT.get(wildcards.sample) threads: 8 output: "03-stacks-analysis/ref-based/{sample}.snps.tsv.gz", "03-stacks-analysis/ref-based/{sample}.tags.tsv.gz", "03-stacks-analysis/ref-based/{sample}.models.tsv.gz", "03-stacks-analysis/ref-based/{sample}.alleles.tsv.gz" log: "03-stacks-analysis/ref-based/{sample}.pstacks.oe" shell:""" mkdir -p 03-stacks-analysis/ref-based pstacks -p {threads} -t bam -f {input} -i {params.index} -o {params.outdir} &> {log} """ ## A catalog can be built from any set of samples processed by the ustacks or pstacks programs rule ref_based_cstacks: input: "info/popmap.tsv",expand("03-stacks-analysis/ref-based/{sample}.snps.tsv.gz", sample=SAMPLES) threads: 10 output: "03-stacks-analysis/ref-based/batch_1.catalog.alleles.tsv.gz", "03-stacks-analysis/ref-based/batch_1.catalog.snps.tsv.gz", "03-stacks-analysis/ref-based/batch_1.catalog.tags.tsv.gz" shell: "cstacks -p {threads} --aligned -P 03-stacks-analysis/ref-based/ -M {input[0]}" ## Sets of stacks, i.e. putative loci, constructed by the ustacks or pstacks programs ## can be searched against a catalog produced by cstacks. rule ref_based_sstacks: input: "03-stacks-analysis/ref-based/{sample}.snps.tsv.gz", "03-stacks-analysis/ref-based/{sample}.tags.tsv.gz", "03-stacks-analysis/ref-based/{sample}.models.tsv.gz", "03-stacks-analysis/ref-based/{sample}.alleles.tsv.gz" params: catalog = "03-stacks-analysis/ref-based/batch_1", sample = lambda wildcards: "03-stacks-analysis/ref-based/" wildcards.sample output: "03-stacks-analysis/ref-based/{sample}.matches.tsv.gz" log: "03-stacks-analysis/ref-based/{sample}.sstacks.oe" shell:""" sstacks --aligned -c {params.catalog} -s {params.sample} -o 03-stacks-analysis/ref-based &> {log} """将以上代码保存为Snakefile,没有集群服务器,就直接用 snakemake运行吧。因为我能在集群服务器上提交任务,所以用如下代码 snakemake --cluster "qsub -V -cwd" -j 20 --local-cores 10 &plot_n_snps_per_locus.R的代码 #!/usr/bin/env Rscript snps_per_loc = read.delim(\'./n_snps_per_locus.tsv\') # Keep only M==n, m==3 snps_per_loc = subset(snps_per_loc, M==n & m==3) # Rename column 1 colnames(snps_per_loc)[1] = \'par_set\' # Create a new data frame to contain the number of loci and polymorphic loci d = snps_per_loc[,c(\'par_set\', \'M\', \'n\', \'m\')] d = d[!duplicated(d),] # Compute these numbers for each parameter set, using the par_set column as an ID rownames(d) = d$par_set for(p in rownames(d)) { s = subset(snps_per_loc, par_set == p) d[p,\'n_loci\'] = sum(s$n_loci) s2 = subset(s, n_snps > 0) d[p,\'n_loci_poly\'] = sum(s2$n_loci) } # Make sure the table is ordered d = d[order(d$M),] pdf(\'./n_loci_Mn.pdf\') # Number of loci # ========== plot(NULL, xlim=range(d$M), ylim=range(c(0, d$n_loci)), xlab=\'M==n\', ylab=\'Number of loci\', main=\'Number of 80%-samples loci as M=n increases\', xaxt=\'n\', las=2 ) abline(h=0:20*5000, lty=\'dotted\', col=\'grey50\') axis(1, at=c(1,3,5,7,9)) legend(\'bottomright\', c(\'All loci\', \'Polymorphic loci\'), lty=c(\'solid\', \'dashed\')) lines(d$M, d$n_loci) points(d$M, d$n_loci, cex=0.5) lines(d$M, d$n_loci_poly, lty=\'dashed\') points(d$M, d$n_loci_poly, cex=0.5) # Number of new loci at each step (slope of the previous) # ========== # Record the number of new loci at each parameter step d$new_loci = d$n_loci - c(NA, d$n_loci)[1:nrow(d)] d$new_loci_poly = d$n_loci_poly - c(NA, d$n_loci_poly)[1:nrow(d)] # Record the step size d$step_size = d$M - c(NA, d$M)[1:(nrow(d))] plot(NULL, xlim=range(d$M), ylim=range(c(0, d$new_loci, d$new_loci_poly), na.rm=T), xlab=\'M==n\', ylab=\'Number of new loci / step_size (slope)\', main=\'Number of new 80%-samples loci as M=n increases\' ) abline(h=0, lty=\'dotted\', col=\'grey50\') legend(\'topright\', c(\'All loci\', \'Polymorphic loci\'), lty=c(\'solid\', \'dashed\')) lines(d$M, d$new_loci / d$step_size) points(d$M, d$new_loci / d$step_size, cex=0.5) lines(d$M, d$new_loci_poly / d$step_size, lty=\'dashed\') points(d$M, d$new_loci_poly / d$step_size, cex=0.5) null=dev.off() plot_n_loci.R的代码 #!/usr/bin/env Rscript d = read.delim(\'./n_snps_per_locus.tsv\') # Keep only M==n, m==3. d = subset(d, M==n & m==3) # Make sure the table is ordered by number of snps. d = d[order(d$n_snps),] Mn_values = sort(unique(d$M)) # Write the counts in a matrix. m = matrix(NA, nrow=length(Mn_values), ncol=max(d$n_snps) 1) for(i in 1:nrow(d)) { m[d$M[i],d$n_snps[i] 1] = d$n_loci[i] # [n_snps 1] as column 1 is for loci with 0 SNPs } # Truncate the distributions. max_n_snps = 10 m[,max_n_snps 2] = rowSums(m[,(max_n_snps 2):ncol(m)], na.rm=T) m = m[,1:(max_n_snps 2)] m = m / rowSums(m, na.rm=T) # Draw the barplot. pdf(\'n_snps_per_locus.pdf\') clr = rev(heat.colors(length(Mn_values))) barplot(m, beside=T, col=clr, las=1, names.arg=c(0:max_n_snps, paste(\'>\', max_n_snps, sep=\'\')), xlab=\'Number of SNPs\', ylab=\'Percentage of loci\', main=\'Distributions of the number of SNPs per locus\nfor a range of M==n values\' ) legend(\'topright\', legend=c(\'M==n\', Mn_values), fill=c(NA, clr)) null=dev.off()第三步:过滤并导出数据 这一步的过滤在stacks1.47是分为两个部分。第一部分是对于从头分析和基于参考基因组都使用rxstacks过滤掉低质量变异,然后重新用cstacks和sstacks处理。第二部分是使用population从群体角度进行过滤。 在stacks2.0时代,rxstacks功能不见了(我推测是作者认为作用不大),既然他们都不要了,那我也就只用population过滤和导出数据了。 这一步主要淘汰那些生物学上不太合理,统计学上不显著的位点,一共有四个主要参数负责控制,前两个是生物学控制,根据研究主题而定 (-r):该位点在单个群体的所有个体中的最低比例(-p): 该位点至少需要在几个群体中存在(—min_maf): 过滤过低频率位点,推荐5~10%(—max_obs_het): 过滤过高杂合率位点, 推荐60~70%# 项目根目录 min_samples=0.80min_maf=0.05max_obs_het=0.70populations -P 03-stacks-analysis/ref-based/ -r $min_samples --min_maf $min_maf \ --max_obs_het $max_obs_het --genepop &> populations.oe # --genepop表示输出格式,可以选择--vcf等最后会在03-stacks-analysis/ref-based/生成至少如下几个文件: batch_1.sumstats.tsv: 核酸水平上的描述性统计值,如观测值和期望的杂合度 π, and FISbatch_1.sumstats_summary.tsv: 每个群体的均值batch_1.hapstats.tsv:单倍型水平的统计值,如基因多样性和单倍型多样性batch_1.haplotypes.tsv: 单倍型batch_1.genepop:指定的输出格式batch_1.populations.log:输出日志至此,上游分析结束,后续就是下游分析。后续更新计划: stacks总体流程鸟瞰Stacks核心参数深入了解RAD-seq和GBS技术比较不同简化基因组protocol的比较学习TASSEL-GBS数据分析流程下游分析探索:这个得慢慢来
|
【本文地址】