| 一种高纯度二氧化双环戊二烯DCPDDO的制备方法与流程 | 您所在的位置:网站首页 › 双环戊二烯工艺流程图 › 一种高纯度二氧化双环戊二烯DCPDDO的制备方法与流程 |
一种高纯度二氧化双环戊二烯DCPDDO的制备方法与流程
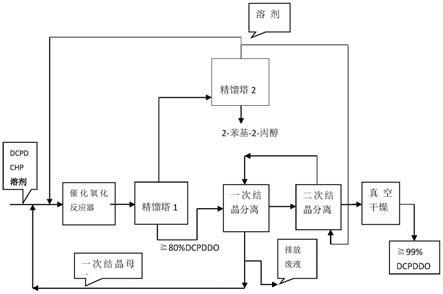 一种高纯度二氧化双环戊二烯dcpddo的制备方法技术领域1.本发明属于催化氧化合成有机环氧化合物及烯烃催化氧化技术领域,具体的说,涉及一种以双环戊二烯为原料,钛硅分子筛为催化剂,制备高纯度(3,4),(8,9)-双环氧-三环[5,2,1,02,6]癸烷(俗称二氧化双环戊二烯dcpddo)的方法。 一种高纯度二氧化双环戊二烯dcpddo的制备方法技术领域1.本发明属于催化氧化合成有机环氧化合物及烯烃催化氧化技术领域,具体的说,涉及一种以双环戊二烯为原料,钛硅分子筛为催化剂,制备高纯度(3,4),(8,9)-双环氧-三环[5,2,1,02,6]癸烷(俗称二氧化双环戊二烯dcpddo)的方法。背景技术: [0002]双环戊二烯(dcpd),学名:三环[5,2,1,02,6]癸二烯-3,8,是石油裂解碳五馏分中的一种重要组分,约占碳五馏分的14%~19%。双环戊二烯的环氧化反应产物二氧化双环戊二烯(dcpddo),学名(3,4),(8,9)-双环氧-三环[5,2,1,02,6]癸烷,是一种性能优异的脂环族环氧化物。相比于普通环氧树脂,二氧化双环戊二烯在耐高温、耐热性、耐候性、耐紫外线、电绝缘、高强度等方面有着更加良好的表现。基于以上性质,二氧化双环戊二烯被广泛用于耐高温浇注料、玻璃钢、粘合剂、层压材料及电子器件封装等方面。[0003]现有技术中,二氧化双环戊二烯通常采用过氧乙酸法、氯醇法和氢化过氧化物催化环氧化法等方法由双环戊二烯经环氧化反应制得,但目前这三种方法均存在不足之处,比如反应工艺复杂、设备腐蚀严重、容易导致环氧化物的酸性开环生成副产物、“三废”排放量多等缺点。近年来,以双氧水为氧源、杂多酸类化合物为催化剂的绿色环氧化工艺受到了广泛关注。根据催化作用机理可以将催化剂分为均相催化剂和非均相催化剂,均相催化反应是直接将杂多酸或其盐类添加到反应体系中,催化效率高,但催化剂不易回收,成本高且“三废”多;而非均相催化反应在保持相对较高的催化效率的同时也实现了对催化剂的回收利用,因而得到了国内外科研工作者的广泛关注和深入研究。venturello等人报道了na2wo4/h3po4/h2o2两相催化环氧化反应体系,在相转移催化剂存在下,反应对环己烯、苯乙烯等大部分烯烃转化率在95%,环氧化合物选择性在80%左右(j.org.chem,1983,48(21):3831-3833)。ishii等人报道了杂多酸h3pw12o40或h3pmo12o40与十六烷基氯化吡啶组成催化剂存在下,用35%的h2o2对多种有机底物进行环氧化反应,在均相或两相体系都能有效进行。但该催化剂体系用于二氧化双环戊二烯合成时,存在杂多酸易流失,催化剂分离回收难度大、重复使用次数少等缺点,难以用于工业化装置(j.org.chem,1998,53(15):3587-3593)。李丽等人报道了将h3pw12o40浸渍二氧化硅表面,以h2o2为氧化剂,用于二氧化双环戊二烯合成,催化剂初活性很高,但在反应过程中,吸附于二氧化硅表面的杂多酸容易流失,催化剂不能重复使用(杂多酸催化双环戊二烯制备二氧化双环戊二烯的绿色合成新工艺[d].吉林,东北师范大学,2007.)。由此可见,通过浸渍法、溶胶-凝胶法将杂多酸负载于二氧化硅表面,虽然可以解决催化剂回收问题,但杂多酸在反应过程中容易从催化剂载体表面脱落,导致催化剂寿命短,一般重复使用5~6次后,催化剂活性显著下降,不易用于工业生成装置。综上,通过非均相催化氧化法制备二氧化环氧戊二烯,虽然实现了对催化剂的回收利用,但存在杂多酸易于脱落导致催化剂寿命短的问题。[0004]由于dcpd分子存在两个双键,因此为了使dcpd完全转化为二氧化双环戊二烯,需用过量的有机过氧化物。而过量的有机过氧化物对产物分离提纯带来许多问题,对催化剂活性也带来许多不利影响。如果过氧化物不过量,部分dcpd仅一个双键发生环氧化反应,生成单氧化双环戊二烯,有两种分子结构,学名分别为3,4-环氧基-三环[5,2,1,02,6]癸烯-8,或8,9-环氧基-三环[5,2,1,02,6]癸烯-3,作为化学品中间体,在精细化工、环氧树脂领域有重要应用前景。 技术实现要素: [0005]为了解决二氧化双环戊二烯合成工艺的缺陷,发明人发现,采用钛硅分子筛催化剂,有机过氧化物为氧化剂,可以将双环戊二烯有效合成二氧化双环戊二烯(dcpddo),反应收率可以到95%~100%。现有的ti-hms分子筛催化剂用于dcpddo合成,初活性好,但随着使用次数增加,反应活性显著下降。重要原因是由于过量的有机过氧化物会给反应系统引入更多的水,反应过程中有机过氧化物自身部分也分解会产生水和酸,易造成反应产物自身聚合,引起催化剂孔道堵塞,hms骨架破裂、钛流失,从而使催化剂失活,这就限制了ti-hms用于二氧化双环戊二烯合成。[0006]为了解决这一问题,首先,本发明对具有介孔特征的含钛分子筛催化剂ti-hms进行改进,催化剂中引入氧化镁、纳米碳纤维,使ti-hms防水、抗结焦性能有了很大的改善。其次,通过控制反应原料配比,确保在反应器中的dcpd、有机过氧化物转化率大于99%。再次,利用精馏分离和结晶分离技术,分离出二氧化双环戊二烯,产物中的单氧化双环戊二烯,或未反应的chp、dcpd原料重新回收到反应器,与新鲜反应原料混合后继续反应,有效解决了反应产物中过量有机过氧化物造成的产品分离提纯等问题。[0007]本发明的技术方案具体介绍如下。[0008]本发明提供一种高纯度二氧化双环戊二烯dcpddo的制备方法,包括:[0009](1)先以双环戊二烯dcpd为反应原料,过氧化氢异丙苯chp为氧化剂,在惰性溶剂中,dcpd、chp与钛硅分子筛催化剂充分接触下发生催化氧化反应,生成主产物(3,4),(8,9)-双环氧-三环[5,2,1,02,6]癸烷dcpddo和2-苯基-2-丙醇的混合液;[0010]其中,所述钛硅分子筛催化剂为具有介孔特征的改性ti-hms分子筛催化剂,其组分包括:tio2质量分数为0.10%~6.42%,纳米碳纤维质量分数为44.68%~64.11%,sio2质量分数为28.17%~45.80%,氧化镁质量分数为0.01%~1.98%,硅烷基质量分数为0.44%~6.93%,所述催化剂uv-vis谱图在220nm处有很强的吸收峰;[0011]催化氧化反应温度为30~150℃、压力0~10mpa(表压),dcpd与chp摩尔比为1.0:1.8~1.0:2.5,dcpd与惰性溶剂体积比为1:4~1:30;[0012](2)将步骤(1)得到的混合液输送到第一精馏塔,进行单塔减压精馏分离,塔顶收集惰性溶剂、2-苯基-2-丙醇混合物,塔釜收集dcpddo物料;塔顶操作压力优选300~5000帕,回流比10:1~1:5,塔顶收集馏分温度15~90℃,塔釜物料温度为90~155℃;[0013](3)取步骤(2)精馏塔釜收集的dcpddo物料,加入惰性有机溶剂配成dcpddo饱和溶液,dcpddo与惰性有机溶剂质量比为1:2~1:20,在60~100℃温度下配置成dcpddo和溶液,将所述饱和溶液冷却到-5~30℃,溶液中析出dcpddo固体,一次结晶分离得到固体dcpddo和结晶母液;[0014]将分离得到得固体dcpddo重复步骤(3)结晶分离操作,用新鲜惰性有机溶剂进行重结晶分离提纯,得到二次结晶分离出的固体dcpddo和二次结晶母液,二次结晶分离得到的结晶母液,作为一次结晶分离的溶剂,保存备用;[0015](4)将步骤(3)二次结晶分离得到的固体dcpddo在压力100~3000pa,温度50~100℃环境下,干燥0.5~5小时,得到质量分数≧99%的dcpddo;[0016](5)将步骤(2)精馏塔塔顶得到的溶剂和2-苯基-2-丙醇混合物输送到第二精馏塔,进行减压精馏分离操作,塔顶收集溶剂,塔釜收集2-苯基-2-丙醇,塔顶收集的溶剂作为反应溶剂和dcpddo二次结晶分离的溶剂。[0017]进一步的,所述步骤(1)中的催化氧化反应温度为60~120℃、压力为0.3~3mpa(表压),chp与dcpd摩尔比为1.8:1~2.2:1。[0018]进一步的,所述步骤(1)中的催化氧化反应采用间歇式反应工艺或固定床连续化反应工艺;采用间歇式反应工艺,催化剂用量与dcpd质量比为0.01:100~20:100,反应时间为1~20小时;采用固定床连续化反应工艺,总物料体积空速为0.1~4.2h-1。[0019]进一步的,所述步骤(1)中的催化剂与dcpd的质量比为0.5:100~10:100,反应时间为3~6小时。[0020]进一步的,所述步骤(1)中的惰性溶剂选自己烷、庚烷、辛烷、壬烷、癸烷、十一烷、苯、甲苯、乙苯、甲乙苯、二甲苯、异丙苯、石油醚中任意一种。[0021]进一步的,所述步骤(2)中塔顶操作压力500~2000帕,回流比5:1~1:2,塔顶温度40~90℃,塔釜温度90~130℃。[0022]进一步的,所述步骤(3)中的dcpddo与所述有机溶剂质量比1:4~1:20,所述饱和溶液冷却至20~30℃。[0023]进一步的,步骤(3)所述的有机溶剂选自己烷、庚烷、辛烷、壬万、癸烷、十一烷、苯、甲苯、乙苯、甲乙苯、二甲苯、异丙苯、石油醚中任意一种。[0024]进一步的,所述步骤(3)中二次结晶分离得到的结晶母液,作为一次结晶分离dcpddo的有机溶剂,回用于粗dcpddo饱和溶液的配置,具体方法如下:(a)取步骤(2)精馏塔釜收集的dcpddo物料与步骤(3)得到的dcpddo二次结晶分离的母液混合,dcpddo和二次结晶母液质量比为1:4~1:20,在60~100℃配成饱和溶液,然后将dcpddo饱和溶液冷却到20~30℃,饱和溶液中析出固体dcpddo,(b)固液分离,得到一次结晶分离的固体dcpddo和一次结晶母液。(c)一次结晶分离得到dcpddo按照步骤(3)给定的结晶分离工艺条件,用新鲜溶剂进行二次结晶分离。[0025]进一步的,所述步骤(3)还包括:[0026]将一次结晶母液送回至步骤(1)的催化氧化反应器,与反应原料dcpd、chp以及惰性溶剂混合继续反应;或输送到步骤(2)的减压精馏塔,精馏分离出结晶母液中的溶剂和dcpddo。[0027]进一步的,所述步骤(5)还包括:[0028]将第二精馏塔顶得到的惰性溶剂中的50%~80%送回至步骤(1)的催化氧化反应器进料口,与反应原料dcpd、chp混合进入反应器反应,重复使用;其余部分的惰性溶剂输送到步骤(3),用作为dcpddo二次结晶分离所需的新鲜溶剂。[0029]进一步的,所述步骤(5)中的减压精馏分离操作,塔顶操作压力300~5000帕,回流比3:1~10:1,塔顶馏分温度15~90℃,塔釜物料温度为85~150℃。[0030]进一步的,所述步骤(5)的塔顶操作压力300~2000帕,回流比3:1~6:1,塔顶馏分温度45~80℃,塔釜物料温度为85~140℃。[0031]上述所提及的钛硅分子筛催化剂,其制备方法如下:[0032]a)在惰性气氛下,将硅源、钛源、模板剂溶解在水和有机醇的混合溶剂中,室温搅拌晶化0.5~10小时,加入纳米碳纤维继续搅拌10~72小时,然后于150~200℃晶化1~7天,结晶产物经分离、洗涤和干燥,得到纳米碳纤维负载的ti-hms;[0033]b)将纳米碳纤维负载的ti-hms投入到含mg(oh)2、mgo、mgco3、4mgco3。mg(oh)2.5h2o、白云石(mgco3.caco3)中任意一种或几种混合物的水溶液中,室温搅拌10~48小时后,分离出固体,水洗液至呈中性,干燥,然后在惰性气氛中,于300~1000℃焙烧2~20小时,得到含镁化合物改性、纳米碳纤维为载体的ti-hms催化剂母体;[0034]c)将ti-hms催化剂母体在25~300℃的温度下用有机硅溶液处理0.5~100小时;有机硅的用量为ti-hms催化剂母体重量的10%~70%;然后过滤分离出固体催化剂,用惰性溶剂洗涤,再将固体催化剂于压力0.133~1.33kpa,温度80~200℃的环境中烘烤10~24h,制得含镁化合物改性的ti-hms/纳米碳纤维复合催化剂。[0035]进一步的,步骤a)中,硅源为正硅酸酯或烃基硅酸酯中的一种或两种;钛源为钛酸酯;模板剂为通式rnh2的有机胺,r为6~18个碳原子的链烃基;rnh2和硅源中的si的摩尔比为0.01~0.3:1,硅源中的si与钛源中的ti的摩尔比为5:1~500:1,水与硅源摩尔比为4~20:1,醇和硅源的体积比为1~4:1,纳米碳纤维与硅源的摩尔比为5~20:1。[0036]进一步的,所用的钛酸酯选自选自钛酸四甲酯、钛酸四乙酯、钛酸四丙酯、钛酸四丁酯、钛酸四异丁酯中的任意一种。[0037]进一步的,步骤a)中,所用的纳米碳纤维,是由含碳气源,优选co、ch4、c2h6、c3h8中的任意一种为反应原料,选用周期表第ⅷ族元素,更优选fe、co、ni中任意一种单金属元素或多种元素合金为催化剂,通过含碳气源催化裂解反应形成的纳米碳纤维,纳米碳纤维催化生长温度为550~650℃。[0038]进一步的,步骤b)中,纳米碳纤维负载的ti-hms与含镁化合物水溶液的体积比为1:1~1:3,mgo、mg(oh)2、mgco3、4mgco3。mg(oh)2.5h2o、白云石中任意一种或几种混合物与步骤a)所用的硅源的摩尔比为0.01:100~7:100;焙烧温度为400~800℃,焙烧时间为3~8小时。[0039]进一步的,步骤c)中,所用的有机硅溶液,有机硅选自卤硅烷、硅氮烷或甲硅烷基胺中的任意一种。[0040]进一步的,步骤c)中,有机硅选自三甲基氯硅烷、三乙基氯化硅、六甲基二硅氮烷或n-三甲基硅烷基咪唑中的任意一种。[0041]进一步的,步骤c)中,所用的有机硅溶液,溶剂选自苯、甲苯、异丙苯、乙苯、环己烷、正庚烷、辛烷、十二烷中的任意一种或几种混合物。[0042]进一步的,步骤c)中,用惰性溶剂洗涤,包括:用甲苯、苯或烷烃惰性溶剂对固体催化剂洗涤3次,每次洗涤所用的惰性溶剂体积是固体催化剂体积5~10倍。[0043]与现有技术相比,本发明具有以下优点:[0044]按照本发明提供的制备方法,用于二氧化双环戊二烯合成,催化剂活性好,寿命长,产物收率高,反应物料中的dcpd、chp得到充分利用,有效解决过量chp带来的产品分离问题,工艺控制简便,反应工艺安全性大大提高。通过结晶分离技术,解决了dcpddo与chp、单氧化双环戊二烯、dcpd分离、提纯问题,未反应的单氧化双环戊二烯、dcpd或chp可以循环利用,产物收率更高。附图说明[0045]图1是本发明制备方法的工艺流程图。具体实施例[0046]以下以实施例进一步说明本发明,但本发明并不局限于实施例。本专利说明书中的产物收率定义:[0047]dcpddo反应收率=反应产物中的dcpddo摩尔数/参与反应的dcpd原料摩尔数*100%。[0048]溶剂精馏收率=每小时塔顶收集的溶剂质量/每小时进塔的溶剂质量*100%[0049]2-苯基-2-丙醇精馏收率=每小时塔顶收集的2-苯基-2-丙醇质量/每小时进塔的2-苯基-2-丙醇质量*100%[0050]dcpddo结晶收率=结晶分离得到的dcpddo质量/结晶分离原料中的dcpddo质量*100%[0051]实施例1:制备钛硅分子筛催化剂[0052]在室温和搅拌条件下,反应器中依次加14.6克十六胺加入到110ml水和110毫升乙醇中,搅拌;将溶于40毫升乙醇的65克正硅酸乙酯、0.12克钛酸乙酯加入到上述溶液中,搅拌50分钟,晶化10小时,投入18.7克纳米碳纤维,继续搅拌10~72小时,反应器升温,反应物料在200℃晶1天,过滤得到固体,用乙醇萃取催化剂中的模板剂,然后纯水洗涤,直到淋洗液呈中性。将洗涤后的固体在110℃烘干12小时后,得到纳米碳纤维为载体的ti-hms。[0053]将上述纳米碳纤维为载体的ti-hms固体中加入3倍体积纯水,1.2克mg(oh)2,室温,搅拌48小时,分离出固体,纯水洗涤,直到水洗液呈中性,80℃干燥30小时,然后450℃,氦气氛中焙烧18小时,得到含镁化合物改性、纳米碳纤维为载体的ti-hms;[0054]将上述制备的粉末催化剂样品压片成型后,破碎、筛分,取20~50目的颗粒50克,投入到反应器中,然后再向反应器中加12.6克三甲基氯硅烷、200毫升甲苯,搅拌,反应温度120℃进行硅烷化10小时,终止反应,取出固体催化剂,甲苯淋洗后,在温度80℃,系统压力0.133kpa的真空系统干燥30小时。[0055]实施例2~9,间歇式反应,催化氧化合成主产物dcpddo和2-苯基-2-丙醇混合液[0056]取钛硅分子筛催化剂、过氧化氢异丙苯chp、溶剂按工艺要求投料到500毫升反应器中,反应完成后,精馏分离出溶剂、2-苯基-2-丙醇和dcpddo。结晶分离dcpddo,结晶母液与新鲜反应原料混合投入反应器,继续下一批次的反应。考察温度、压力、物料配比、反应时间对反应产物dcpddo收率的影响,实施例2~9所用的催化剂组成见表1,实验结果见表2。[0057]表1催化剂质量百分比组成[0058][0059]*注:c—表示纳米碳纤维。[0060]表2间歇式合成dcpddo工艺条件[0061][0062]实施例10~17:固定床连续化反应工艺制备dcpddo和2-苯基-2-丙醇混合液[0063]取钛硅分子筛催化剂50克,填装到100毫升等温固定床反应中器,过氧化氢异丙苯为氧化剂,反应料液通过计量泵打入到反应器中。反应产物通过单塔精馏分离,塔顶收集溶剂、2-苯基-2-丙醇,塔釜为dcpddo粗品。结晶分离dcpddo,结晶母液与新鲜反应原物料混合重新进反应器。实施例10~17为固体床连续化反应,所用的催化剂组成见表3,分别考察温度、压力、物料配比、空速对反应产物dcpddo收率的影响,结果见表4。[0064]表3催化质量百分组成[0065][0066]*注:c—表示纳米碳纤维。[0067]表4固定床合成dcpddo工艺条件[0068][0069]实施例18~22:第一精馏塔将溶剂、2-苯基-2-丙醇与dcpddo、未反应原料分离[0070]分别收集实施例13、14、15、16、17的催化氧化反应产物输送到第一精馏塔,进行单塔减压精馏操作,塔顶收集溶剂、2-苯基-2-丙醇混合物,塔釜收集dcpddo,精馏原料中的溶剂,精馏操作塔顶压力、温度、回流比,反应溶剂、2-苯基-2-丙醇精馏收率见表5。[0071]表5第一精馏塔将溶剂、2-苯基-2-丙醇与dcpddo分离的工艺条件[0072][0073][0074]实施例23~27:结晶分离固体dcpddo[0075]分别将实施例18、19、20、21、22精馏塔塔釜得到的dcpddo,加入二次结晶母液,配成dcpddo热饱和溶液,冷却到较低温度,利用结晶分离原理,过滤分离出固体dcpddo和一次结晶母液,一次结晶母液回收,与新鲜的dcpd、chp、溶剂混合,进反应器继续反应。一次结晶得到固体dcpddo用新鲜溶剂进行第二次结晶提纯,二次结晶母液作为一次重结晶分离dcpddo的溶剂,重复使用。结晶分离选用溶剂、物料配比、热饱和溶液温度、物料冷却温度、dcpddo结晶收率见表6。一、二次结晶分离dcpddo操作工艺,见图1。[0076]表6结晶分离dcpddo工艺条件[0077][0078]实施例28~32:dcpddo固体样品提纯后烘干处理[0079]分别将实施例23、24、25、26、27二次结晶分离得到的dcpddo固体样品,分批在不同温度和压力下,烘干处理,考察温度、系统压力和烘干时间对dcpddo质量分数的影响,实验结果见表7。[0080]表7 dcpddo真空干燥工艺条件[0081][0082]实施例33~37:第一精馏塔顶收集的溶剂和2-苯基-2-丙醇混合物,通过第二塔精馏分离,塔顶收集溶剂,塔釜收集2-苯基-2-丙醇[0083]将实施例18、19、20、21、22精馏塔顶收集的溶剂和2-苯基-2-丙醇混合物输送到第二精馏塔,进行减压精馏分离溶剂和2-苯基-2-丙醇,塔顶收集溶剂,塔釜收集2-苯基-2-丙醇,分别考察温度、塔顶压力、釜温、回流比及溶剂精馏收率,实验结果见表8。[0084]表8第二精馏塔精馏分离溶剂、2-苯基-2-丙醇工艺条件[0085][0086]实施例38结晶母液中未反应的chp重复利用[0087]取实施例27的结晶母液784克,投入2l高压釜中,结晶母液质量百分组成:dcpddo质量分数为2.1%,chp质量分数2.0%,2-苯基-2-丙醇质量分数为0.4%,正辛烷96%。然后,继续向高压釜中投入98%的dcpd 71.4克,83%chp176克,实施例5所用的催化剂15克,反应系统用氮气置换后,反应温度控制在80~85℃,压力1mpa,机械搅拌,反应5小时,将料液冷却到室温,分析产物组成,dcpd质量分数为0.1%和单氧化双环戊二烯质量分数为0.2%,dcpddo质量分数为9.8%,dcpddo反应收率为99.3%。将反应液减压精馏分离出正辛烷和2-苯基-2-丙醇,得到粗dcpddo固体108克。取1500毫升具回流冷凝管的三口玻璃烧瓶中,投入108克粗dcpddo和1000克dcpddo二次结晶母液(含正辛烷96%),水浴锅加热到70℃,得到dcpddo饱和溶液,冷却到室温,析出固体dcpddo,过滤分离出固体100克dcpddo。将新鲜正辛烷600克与前述的100克dcpddo投入取1500毫升具回流冷凝管的三口玻璃烧瓶中,水浴锅加热到70℃,再次得到dcpddo饱和溶液,然后冷却到25℃,过滤分离出固体dcpddo,在系统压力为500帕,80~90℃干燥4小时,冷却到室温,得到82.2克质量分数大于99%的dcpddo。[0088]以上已对本发明创造的较佳实施例进行了具体说明,但本发明创造并不限于所述实施例,熟悉本领域的技术人员在不违背本发明创造精神的前提下还可作出种种的等同的变型或替换,这些等同的变型或替换均包含在本技术权利要求所限定的范围内。 |
【本文地址】