| 干货来了 | 您所在的位置:网站首页 › 一期临床试验设计的特殊性 › 干货来了 |
干货来了
|
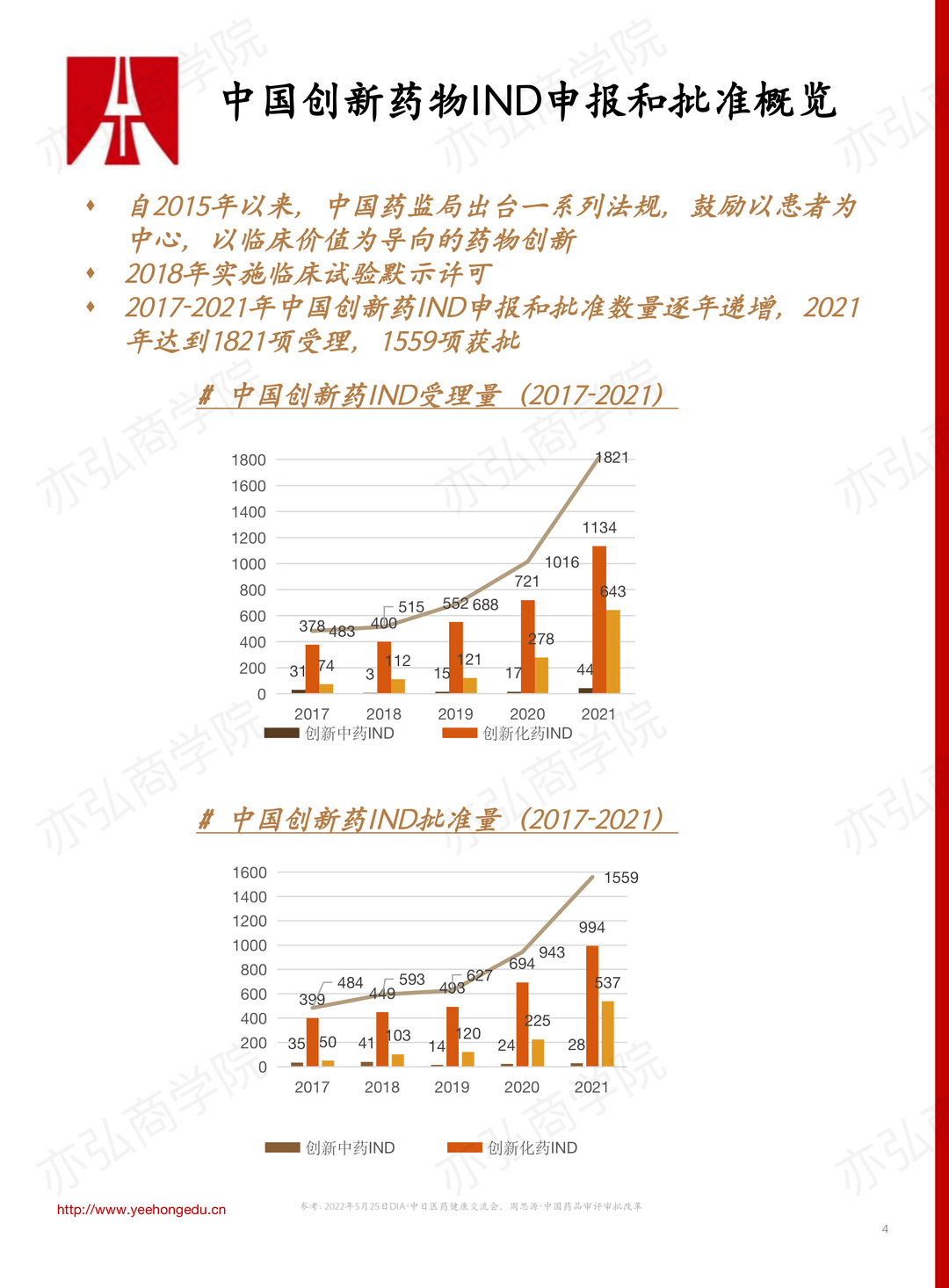
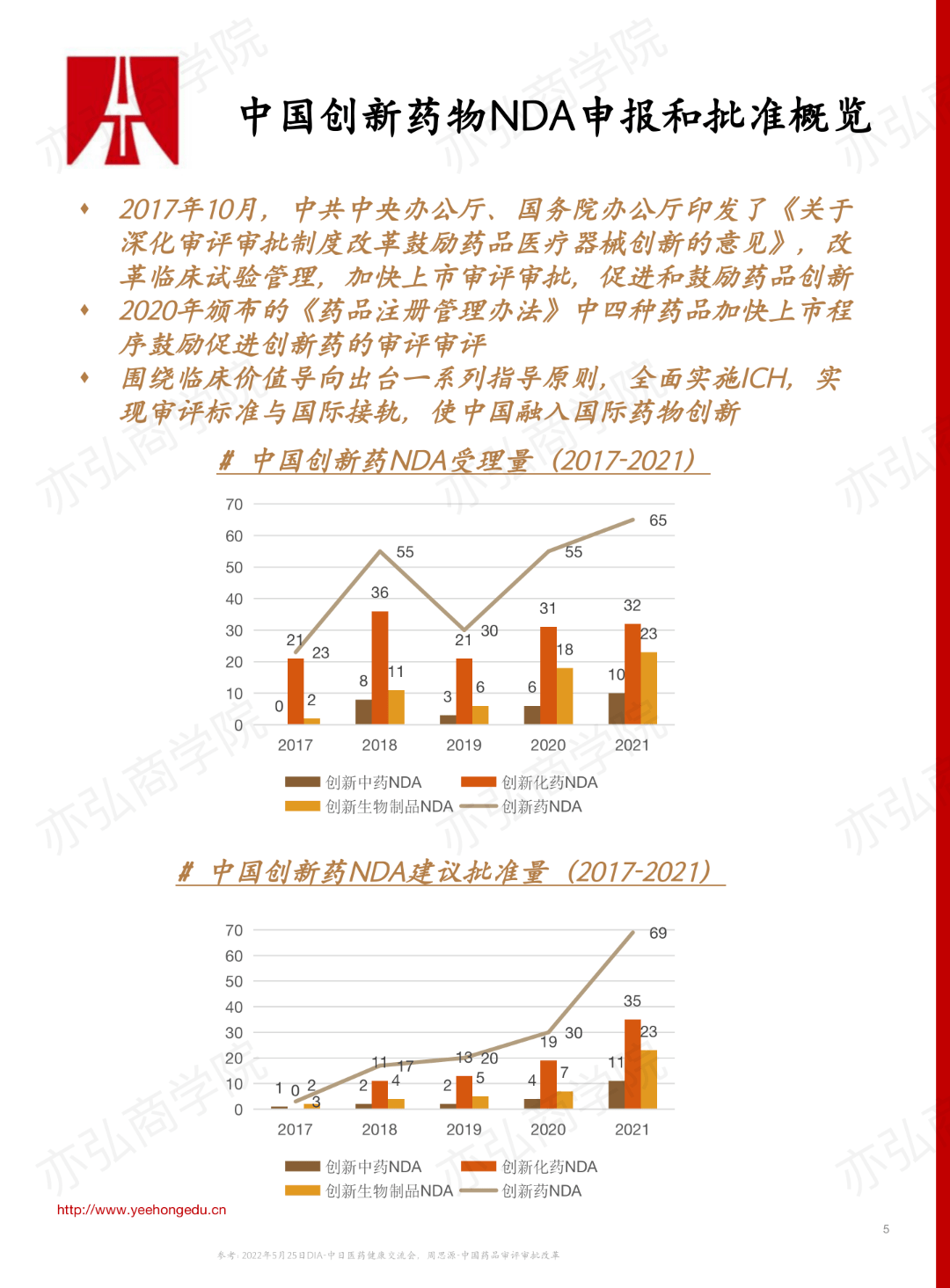
王惠娜老师围绕报告主题,分享了2017年药品法规改革以来创新药临床开发申报的趋势、创新药临床研发过程中Ⅰ期临床试验的内容以及指导原则。 一、创新药临床开发申报的趋势 一系列政策法规的颁布,促进了我国创新药的研发。从2017-2021年,中国创新药临床研究申请(Investigational New Drug,IND)和新药上市申请(New Drug Application,NDA)的申报和批准的数量逐年增加。
二、创新药Ⅰ期临床试验的内容 Ⅰ期临床试验的主要目的是基于已经完成的临床前研究,去探索药物在人体中的适用性,寻找合适的人群、合适的剂量,合适的用药方式和方法。
三、创新药Ⅰ期临床试验指导原则 当前药监局和药品审评中心为促进创新药Ⅰ期临床研究,颁布了一系列指导原则,以指导I期临床研究的设计和开展。
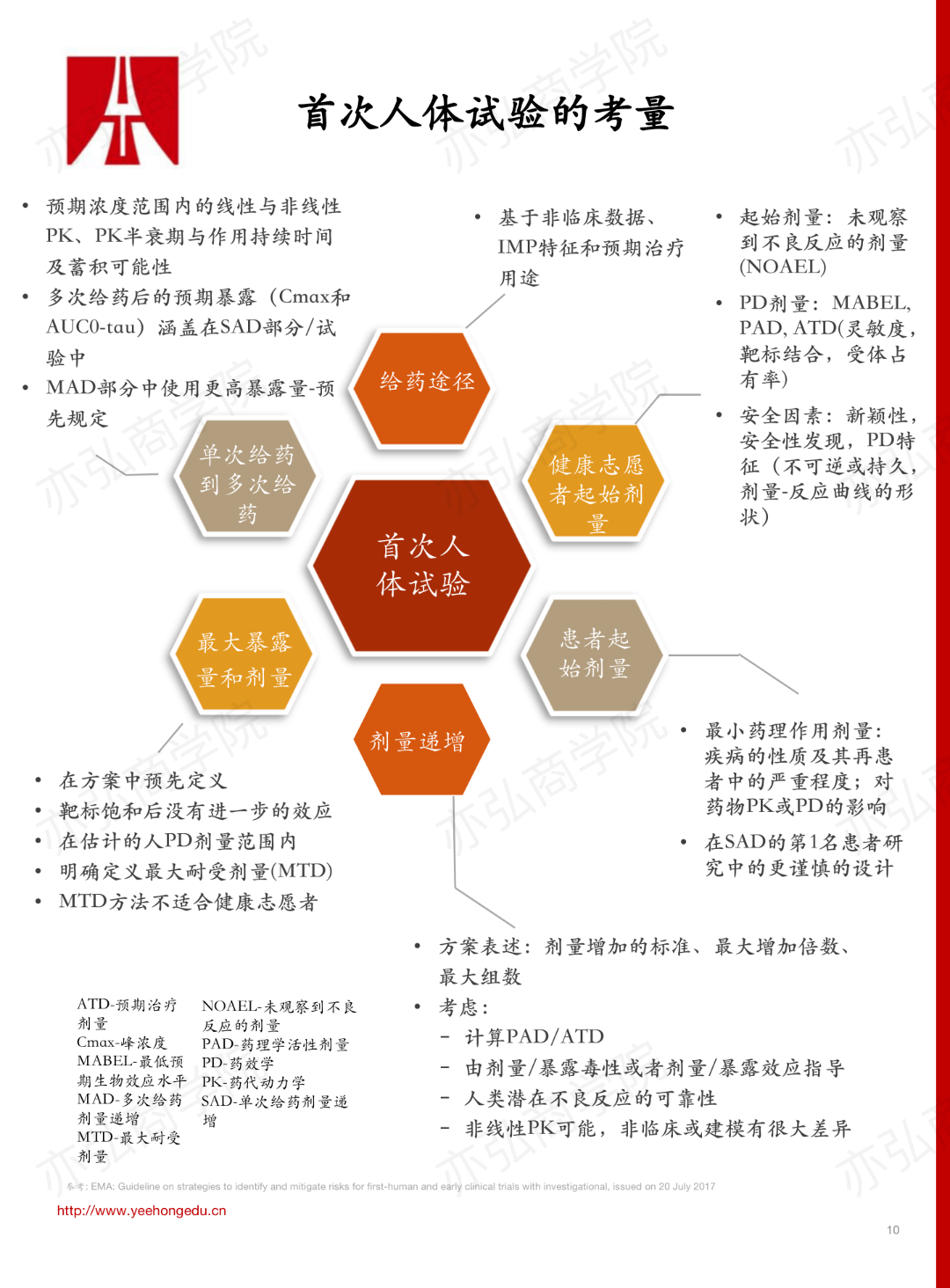
报告中重点分享了首次人体试验的考量、I期临床试验的研究方法、以模型引导(MIDD)为特色的创新药早期开发加速策略以及抗肿瘤药首次人体试验扩展队列研究。 1. 首次人体试验(First in Human,FIH)的考量 对于首次人体试验,要从如下6个方面来进行考量:健康志愿者的起始剂量、患者的起始剂量、剂量递增、最大暴露量和剂量、单次给药到多次给药以及给药途径。
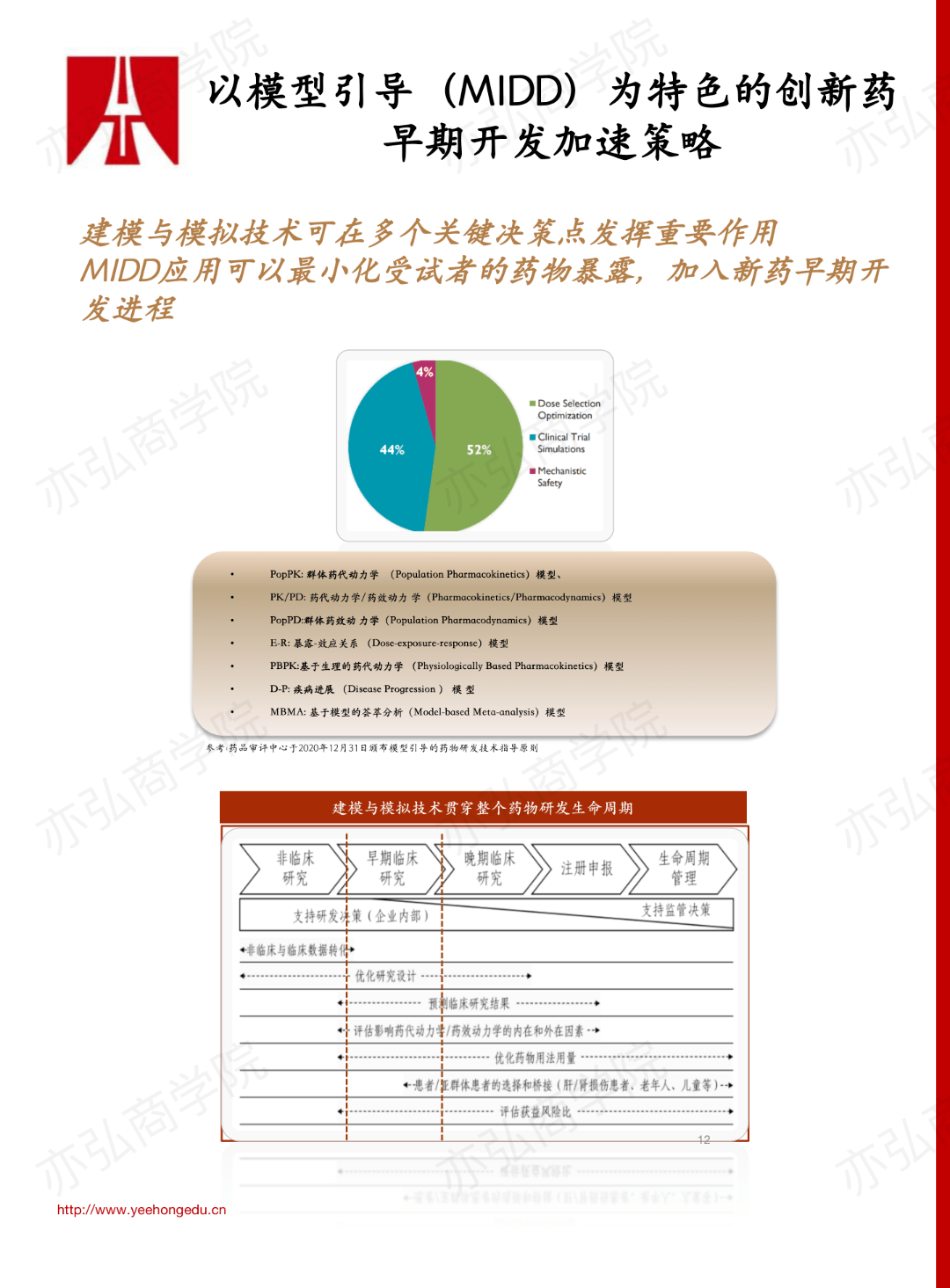
2. I期临床试验的研究方法包括经典研究方法、基于模型的研究方法和其他的前沿方法。其中,基于模型的研究方法是最常用的。 3. 以模型引导(MIDD)为特色的创新药早期开发加速策略 基于模型引导的药物开发贯穿了整个研发周期,涉及优化研究设计、预测临床研究结果、评估药代动力学、药效动力学的内因和外因、优化剂量、人群的选择和评估获益风险,在各个研发节点均发挥着重要作用。模型引导早在I期临床试验中就开始发挥作用,而且是使用最多的。总体来说,MIDD应用可以最小化受试者的药物暴露,加速新药早期开发进程。
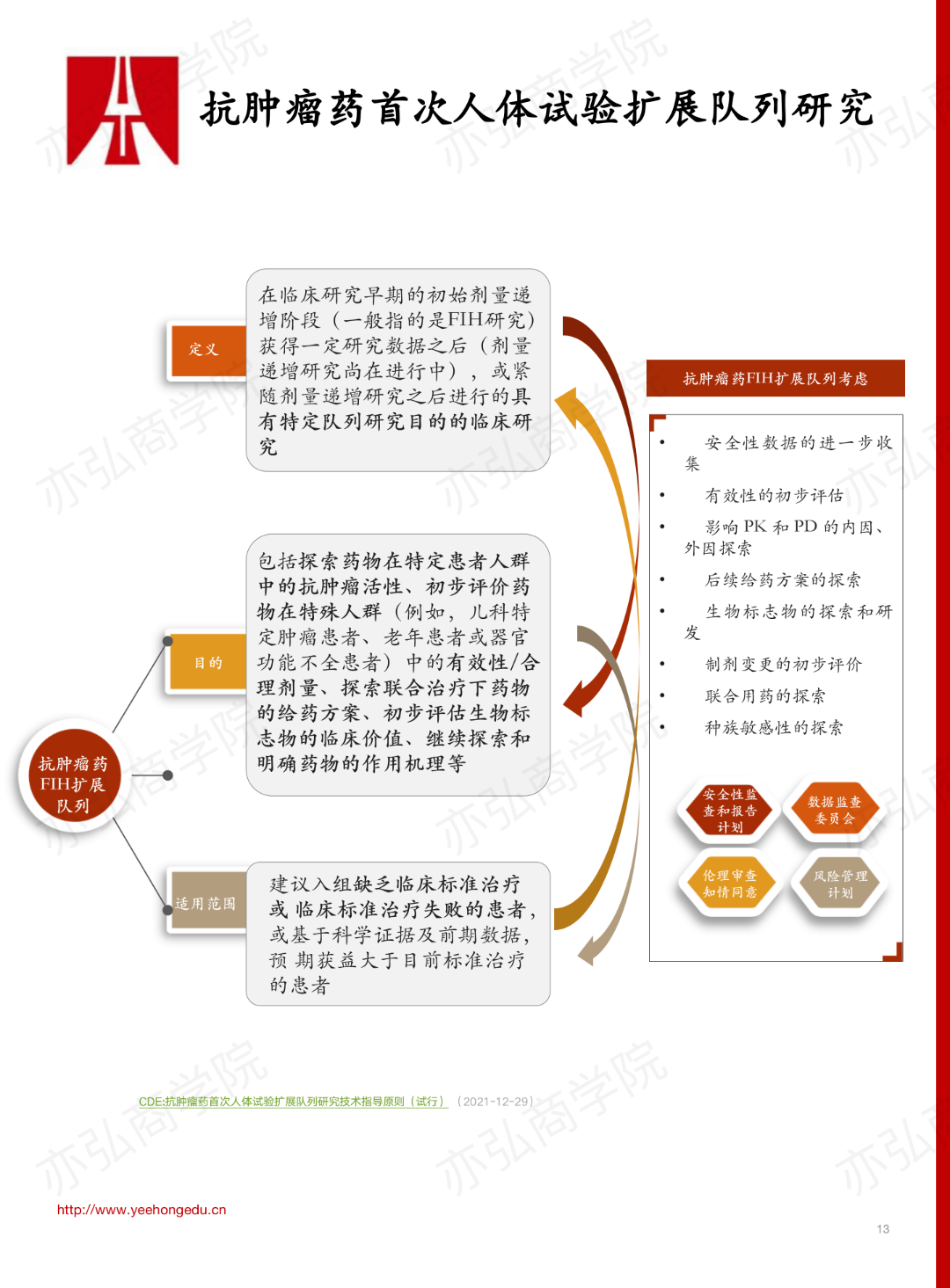
4. 抗肿瘤药首次人体试验扩展队列研究 针对抗肿瘤药物首次人体试验扩展队列研究,CDE在2021年专门颁布了《抗肿瘤药首次人体试验扩展队列研究技术指导原则(试行)》。
总的来说,临床I期试验设计需结合产品自身特点、拟开发适应症、临床治疗领域的特点和临床需求,设计相应的研发计划,提高研发效率。除了传统的I期临床试验,出现了越来越多借助新的工具、模型和试验设计的研究方法,以加速创新药开发,尤其是肿瘤领域创新药的开发。 主旨报告 临床药理学和定量药理学在Ⅰ期研究中的作用
报告嘉宾 纪佳 杨森(中国)研发中心 临床药理和定量药理部门中国团队负责人 纪佳博士从如下三方面进行了分享:临床药理学和定量药理学的定义、Ia期和Ib期研究以及临床药理学研究的应用。 一、临床药理学和定量药理学的定义 临床药理学可以理解为将临床与药理学结合起来。用一个公式可以更好地解释临床药理学,即临床药理学=疾病进展(临床)+药物作用(药理学)。临床药理学贯穿于整个药物研发过程,运用药代动力学(Pharmacokinetics,PK)/药效学(Pharmacodynamics, PD)的机制,支持着研发过程中各阶段主要方案的决策。而定量药理学(也称为群体PK/PD)是将药理学和疾病定量化,从而影响药物研发和监管决策。临床药理学其实是涵盖了定量药理学的。
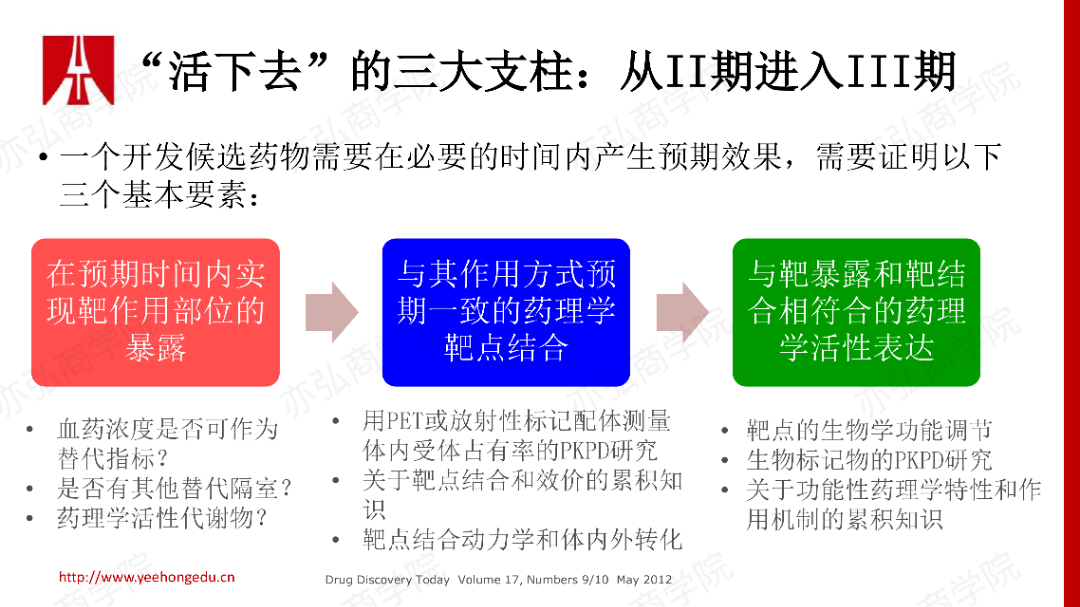
二、Ia期研究和Ib期研究 1. 从II期成功进入到III期的三个基本要素
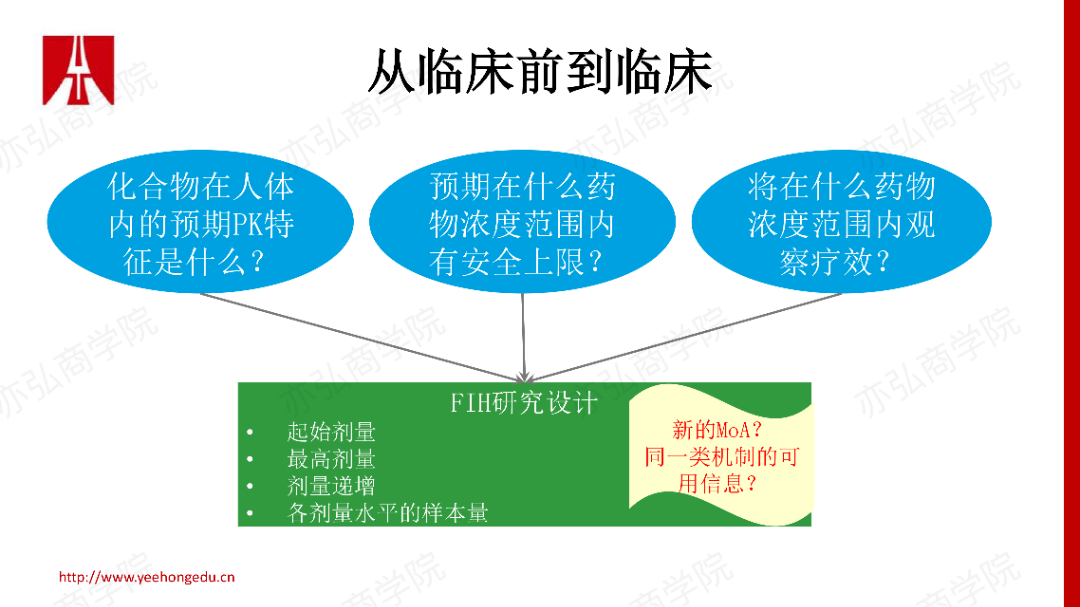
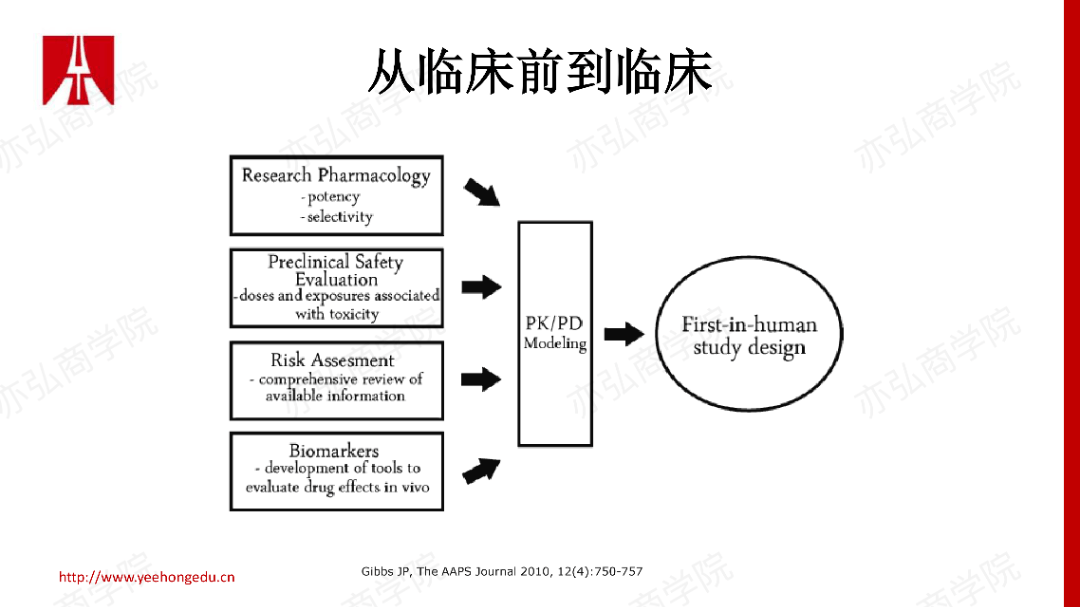
从一篇早期的文献来看,II期研究是失败率最高的阶段。如果一个开发候选药物,它要在必要的时间内产生预期的效果,那么从PK/PD原理方面,我们最看重三个基本要素。 2. 从临床前到临床需要回答的三个问题 我们将如上原理折射到临床前体内体外很多数据的积累,我们来看如何成功地将药物从临床前推到临床研究阶段。在FIH里,我们同样要运用到之前谈到的三大基本要素。在FIH设计前,试图回答三个关键问题。
基于上述三个问题的答案,结合药物是全新的作用机制还是有同类药物,帮助我们更好、更合理地去设计FIH。
3. 定量系统药理学(Quantitative Systems Pharmacology, QSP) QSP可能是定量药理学未来的趋势之一。QSP可以理解为整合体外、靶点、药物生化特性、动物体内试验数据、人体生理病理以及基因组等各方面的信息,试图通过一个非常复杂的数学模型,描述疾病进展和药理作用。
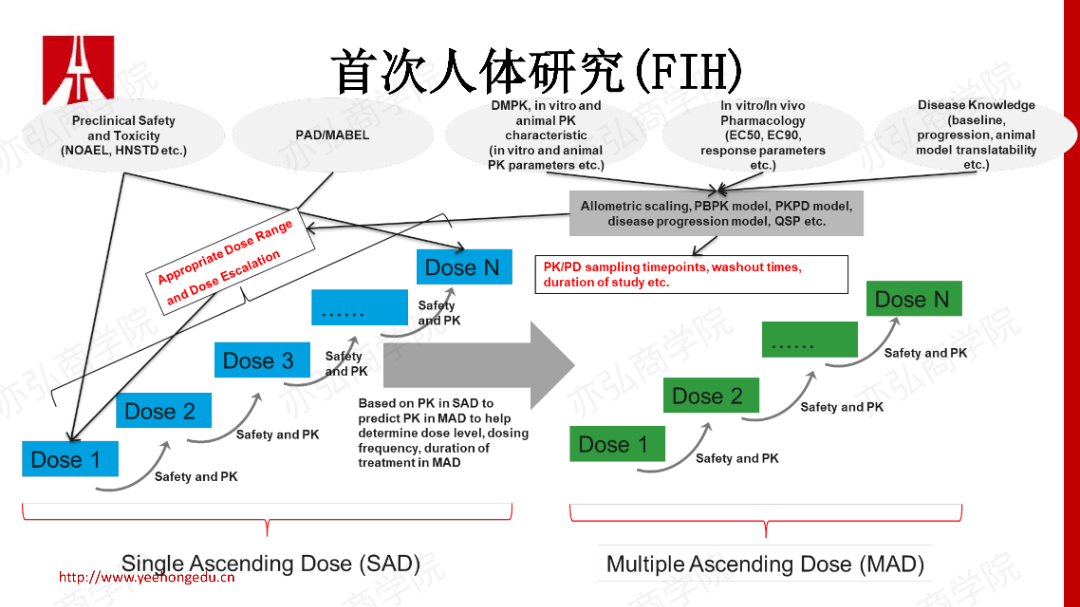
4. FIH 以经典的FIH为例,可以将我们临床前所有的数据整合在模型中,帮助我们更好地研究设计剂量并控制风险。利用单次给药剂量递增(Single Ascending Dose, SAD)的PK数据,去决定多次给药剂量递增(Multiple Ascending Dose,MAD)试验的设计,包括剂量水平、给药频次以及治疗时间。
FIH也会涉及一些其他的队列。
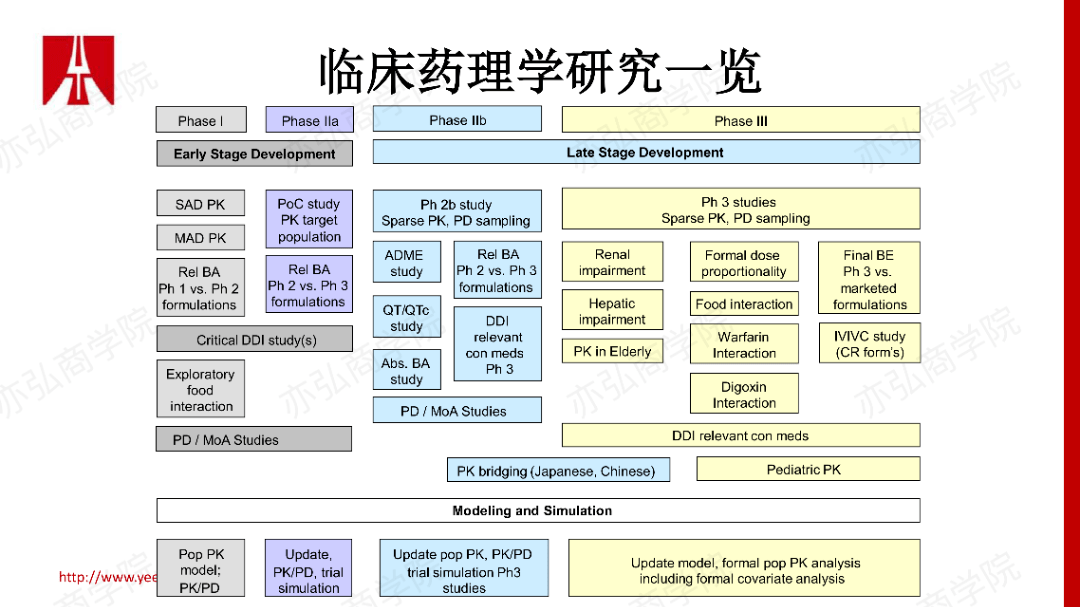
5. Ib期研究 Ib期研究一般是在患者人群中进行,适用于肿瘤和非肿瘤领域的开发。其目的是评估重点给药方案是否能获得目标适应症的早期疗效信号,为概念性验证(Proof of Concept , PoC)二期剂量的确定和剂量扩展阶段提供依据。同时如果目标适应症没有较好的早期疗效,Ib期研究可以发挥早期止损的作用,减少后期研发损耗。Ib期研究患者人群的PK、PD指标的收集非常重要,如生物标记物、探索性终点,早期PK、PD数据收集及建模工作可以更好地支持II期试验推荐剂量(Recommended Phase II dose, RP2D)的确定和PoC的研究设计。 三、临床药理学研究的应用 临床药理学研究作为Ⅰ期研究里很重要的一个分类,其实就是研究内因、外因对体内暴露量的影响,贯穿于药物研发整个过程。根据每个研发阶段的需求,我们会设计相应的、合适的临床药理学研究去支持下一步的决策。
主旨报告 创新药早期研发案例分析与思考
报告嘉宾 李改玲 Certara(上海)医药咨询公司中国区首席科学官,兼全球综合药物开发部高级副总裁 李改玲老师通过对同靶点的两个脂肪酸酰胺水解酶(Fatty Acid Amide Hydrolase,FAAH)抑制剂早期研究案例的对比剖析,分享了临床药理学在转化研究与早期研发中的重要作用。 一、两个案例的剖析 案例1❌:BIA 10-2474 悲剧, 首次人体试验(法国,2016) 在多剂量爬坡到50毫克的第一天,有1位健康受试者死亡。紧接着后面,相继有4位受试者发生严重不良事件(Adverse Event, AE),轰动欧盟监管当局。欧洲药品管理局(European Medicines Agency,EMA)在2017年指南中对I期研究,特别是FIH进行了修改,增加了哨兵法、在健康受试者中无最大耐受剂量(Maximum Tolerated Dose,MTD)的要求。
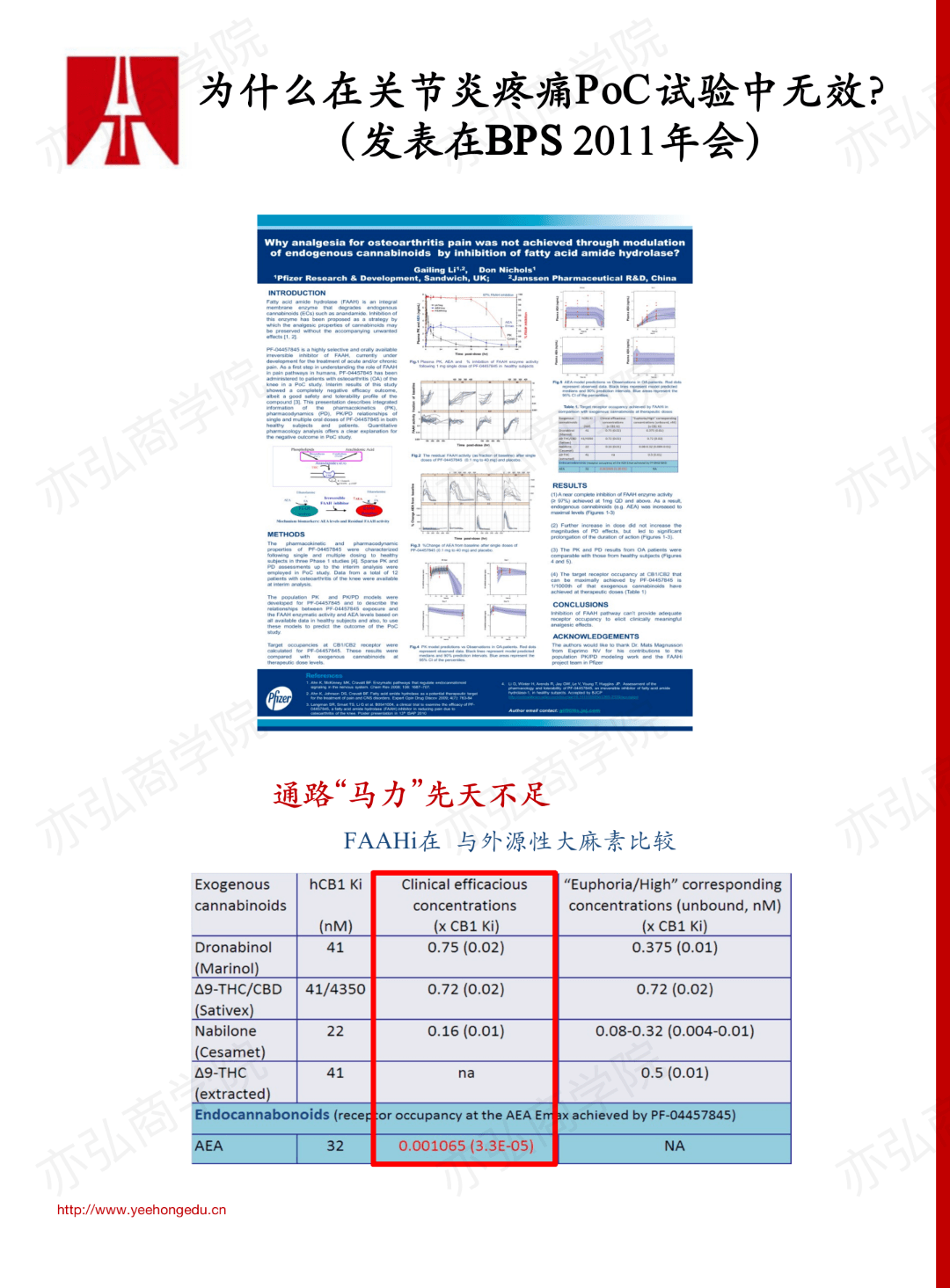
案例2✅:PF-04457845疼痛药研发:FAAH抑制剂
PF-04457845为共价结合FAAH的一种抑制剂,其靶点、生物标志物与活性之间的关系研究表明,需要完全阻断FAAH酶活性 (≥97%)并最大化升高花生四烯乙醇胺(Arachidonoyl Ethanolamide,AEA)水平,才可以在临床前疼痛模型中观察到试验药的抗疼痛作用。临床前研究考察了药物作用随剂量、时间的变化,并评估了药物在外周循环与中枢系统的药代和药效学特征,为临床试验提供了非常详实的证据。 早期临床结果显示:高达40 mg(SD)和8 mg(QD,持续14天)的化合物,在健康人上安全且耐受性非常好,PK表现与非临床所预期的一致;无显著食物影响;在0.5 mg QD剂量下,对FAAH酶活性的持续抑制作用≥97%,FAAs(FAAH 酶底物)持续保持最大升高。但是通过CogState评估未观察到任何中枢系统的应答。当时研究人员对这一现象产生了警惕,并提出质疑:未观察到任何中枢神经系统的效应,那么临床前的药理作用是否能转化为患者的疗效?因此,在后续的PoC研究时, 研究人员慎重设计了一项小型的、在关节炎(Osteoarthropathy,OA)患者中考察疼痛缓解的临床试验。研究结果提示,PF-04457845无效。为什么会这样? 进一步的定量药理学研究发现,与已知外源性大麻素对CB1受体的作用比较,PF-04457845引起的内源性活性物质(AEA等)积累的最大浓度仅为外源性大麻素的几百分之一,这说明该信号通路本身存在不足,其积累的活性成分的量远远不足以有效激活CB1受体及信号通路。
二、造成BIA 10-2474悲剧的原因 通过对BIA 10-2474的研究资料进行梳理,可以得出造成悲剧的7条原因: ① 化合物低效且选择性低,研究不足; ② 报告63页IB仅含少于2页的药理研究,药理学的研究是非常不充分的; ③ 毒理研究未能搞清毒理靶器官和动物死亡原因,为什么采用猴子的毒理实验也未能解释; ④ 前临床到人体研究的转化研究严重不足。在猴子试验上预测人体有效剂量为10-40 mg,而在健康志愿者(Healthy Volunteer,HV)中的数据显示5 mg导致100% FAAH抑制; ⑤ 未重视在首次人体试验中实时检测PK数据; ⑥ 未跟踪检测药物对中枢神经系统的作用,试验中受试者出现视觉模糊的症状,但并未被关注; ⑦ 作用靶点不仅仅是欲设想的单一靶点,严重的脱靶现象是其造成临床悲剧的元凶。 三、是什么导致这两个FAAH抑制剂如此不同? 1. 前临床药理研究严重不足 2. 前临床到人体研究的转化研究严重不足 3. 未能在早期FIH研究中及时发现这些不足,采取相应的措施 主旨报告 创新药Ⅰ期临床研究中安全性评估与决策
报告嘉宾 李海燕 北京大学第三医院药物临床试验机构主任 李海燕老师围绕报告主题,从案例入手,分析了创新药I期临床研究中的风险控制要点,并针对临床上心脏安全性的监测和处理分享了自己的宝贵经验。 一、案例分析—BIA 10-2474悲剧带给我们启示 1. 案例分析 此案例中,有几个关键的失误: 1)在SAD和MAD剂量设置中,剂量的递增幅度不符合常规以及指南。在越接近治疗剂量时,递增的幅度不降反升。 2)研究者在研究过程中,并没有及时地将所有数据放在一起进行分析。虽然从PK的角度,并没有出现明显的异常,但从PD的角度和临床症状上看,已经出现了明显的异常。试验过程中,多处安全性信号均被研究者忽略。 3)在有1名受试者已经送到急诊抢救的情况下,研究者并没有对已经发生的、比较严重的中枢神经系统安全性事件引起足够的重视,非但没有叫停研究,反而给其余的受试者继续给药,最终因严重AE导致试验被迫中止。 2. 启示 1)研究团队须有足够的知识储备,包括对药物生理机制、病理机制、作用机制、种属差异、临床前研究的启示等有充分的了解; 2)在临床前向临床转化的过程中、SAD和MAD实施过程中,应用好现有定量药理学技术,并将所有的数据放在一起实时PK/PD分析,以便快速决策; 3)要考虑到循环中和组织中靶点抑制饱和可能不一致; 4)制定好研究的停止标准,以便及时刹车; 5)实时监测、实时分析、实时适当的调整研究方案。 二、风险监控 在风险监控方面,有如下几点需要重点考虑: 1. 团队需要有足够的知识储备、良好的协作以及快速反应
2. 创新药“新”的程度决定了我们关注度的高低和把握度的大小 对创新药安全性的把控基于对它的了解程度。如果是全新靶点的药物,也无同类药上市,则存在很多未知,意味着风险也高。此时,在谨慎的前提下,需要融合非常全面的知识来支撑安全高效的前行。 3. 常见药物毒性的预测、监测和治疗 在早期试验中,我们除了要关注临床前信号,还要关注几大系统的安全信号,比如心血管、肝脏、肾脏、血液系统、神经系统、免疫系统、消化系统等。对这些安全信号,首先,我们是否可以在临床前对其有一定的预测性;第二,我们能否有手段动态地监测到已发生的信号来帮助我们进行决策;第三,提前考虑可能的监测手段,以及能否处理治疗所发生的安全性事件。 4. 创新药首次人体试验的设计和实施 1)试验设计 首次人体试验设计要基于对药物的了解case by case去考虑。对于高风险的药物,在每个剂量递增阶段,需要采取哨兵法。
2)试验实施 对于高风险研究的实施阶段,要求团队具备紧急情况处理的能力和经验,整合各方面数据进行分析,保持一种警觉状态,适时刹车,避免严重事故的发生。此外,对于不同的药物,安全性终止标准是不一样的。
三、心脏安全性 心脏安全性在药物研发中非常受关注。在报告中,主要分享了QT间期延长引起的尖端扭转室速导致患者猝死。主要要点如下: 1. ICH有专门的指南来指导QTc研究, 如2015年ICH E14 Q&A(R3)、2017年C-QTc白皮书、2020年ICH E14/S7B Q&A。 2. C-QTc研究可以在SAD阶段进行。SAD是药物研发过程中唯一可以把剂量推到最高点的机会。因此,要取到剂量最高点时高质量心电图以及对应的血药浓度。采取建模的方式来定量回答是否有QT间期延长。 主旨报告 创新药I期临床试验核查要点分析
报告嘉宾 王洪允 北京市创新药物临床药代药效研究重点实验室副主任、临床药理中心I期实验室负责人 王洪允老师围绕报告的主题,从三个方面进行了分享:我国创新药I期临床研究现状、创新药I期临床研究要点分析以及创新药I期临床核查思考。 一、我国创新药I期临床研究现状 我国创新药I期临床试验具备如下几个基本特征:1)我国临床试验仍处于早期研发阶段;2)从2011-2020年间,新药I期临床试验共计2842项,其中创新药占53%,涉及创新药品种911种;3)从治疗领域来看,我国I期临床试验以抗肿瘤药物占比最大;4)从受试人群上来看,我国I期临床研究主要在在健康志愿者中进行,患者和特殊人群也有涉及;5)FIH和FIC(中国人群首次人体试验)分别占比显著提高,这提示中国临床试验管理正变得越来越标准化和国际化,中国有能力承担药物研发临床试验的第一阶段。6)我国临床试验处在蓬勃发展的时期,I期临床试验的设计和目标已经发生了很大的变化,质量越来越符合国际标准。创新药I期临床试验所呈现的新特征和新发展趋势,对新药I期临床试验的核查也提出了新的要求。二、创新药I期临床核查要点分析1. 临床试验部分现场核查目的及要点 从核查的目的来看,理论上,核查过程中不会涉及过多科学性的问题,更多地在于现场取证。现场核查包括临床试验部分和生物样品分析的部分。
2. 新版核查要点与2015年版核查要点最大的不同在于,确定风险等级。 新版核查要点基于品种因素和研发主体合规因素进行风险等级划分,并采取“动态调整”的原则。对注册核查风险等级总体上采取“风险就高”原则来开展相关核查。在判定原则上,明确了11种“不通过”的情形,并避免一刀切,更加具有可操作性。
三、创新药I期临床核查思考 创新药的I期临床试验和BE试验的核心目标以及要回答的问题均不相同。
在创新药I期临床研究中所采用的新技术、新方法对核查提出了新的挑战。报告中重点分享一下MIDD应用下的核查要点分析以及14C-人体AME研究相关核查要点分析。 1. MIDD应用之下的核查要点分析 MIDD模型药物从几年前的研发、推广,到现阶段的常规应用给核查带来了新的挑战。 从临床药理学的角度,药物的暴露-反应(Exposure-Response,E-R )关系值得特别关注。 但内外因素都会对E-R分析产生影响,模型的应用就有了必要。E-R分析是MIDD中非常重要的一个内容。E指的是暴露量,通过药代动力学获得,比如传统的PK(Pharmacokinetics,药物代谢动力学)密集型采样以及PPK(Population pharmacokinetics,群体药动学)、PBPK(Physiologically Based Pharmacokinetics,生理药代动力学) )。R指效应,可以是生物标志物,替代终点、早期/临床终点。Ⅰ期试验核查的时候应关注替代终点。 对于使用PK和PPK,我国已经正式颁布相关指导原则,在核查方面是没有问题的。然而,PBPK模型比较复杂,胃肠生理、代谢酶、靶组织靶器官等很多因素都至关重要。虽然PBPK并不是个很新的概念,但目前国内缺少很多系统生物学的基础研究数据,所以机制/半机制的PBPK模型的假设远远多于前两个,其稳定性不可避免地受到较大的影响,其验证的思路和PPK是不一样的,这种时候如果涉及到一些E-R分析、临床终点等或想豁免某些研究的时候,就会增加审评的难度。现场核查标准很难确定。另外PBPK模型的软件有很多,后台不对外开放部分内嵌模型的适用性有待进一步研究对核查提出了新的挑战。PBPK领域的知名研究专家Amin Rostami-Hodjegan近期写了一篇文章关于PBPK模型为什么做、什么时候做、做什么、如何做、由谁来做。对PBPK模型的质量控制具有较大的意义。 2. 14C-人体AME研究相关核查要点 FDA最近出台了人体放射性物质(通常为14C或3H)平衡研究的相关指南,业内专家讨论的非常热烈。2013年国内首个标记试验以及安罗替尼、吡咯替尼等耳熟能详的国内重磅药物,也在国内开展了14C-物质平衡研究并获批上市。随着这种研究越来越普及,我们怎么核查此类试验? 首先,我们要了解这种试验的设计:一般是6~8个健康的男性受试者,口服溶液剂量为100 µCi,然后取尿和粪便。一般会涉及到药理学剂量的设计、放射性剂量的设计、药物的制剂设计(如何标记)、受试者的选择、样本采集、放射性分析等。据了解这种放射性是很安全的,操作的时候戴个手套就可以了。 其次,在核查的时候要关注以下几点: 第一,开展放射性同位素临床试验的单位和人员必须获得相应的资质,并满足硬件、软件和人员的相应要求。此外,要关注特殊性试验细节,比如试验放射性和非放射性废弃物的处理方式。 第二,相关样本的检测。首先要了解检测内容,比如不同时间点全血的总放射量的测定,排泄物的放射量的测定等。除此之外,最终的物料平衡研究通过定性定量的分析,比如代谢谱、结构鉴定等实现相应的测定。
-to be continued- 相关链接: 主席招募 | 亦弘期待和您共赴星辰大海 直播回放|创新药I期临床研究技术指南研讨会 点 击 图 片 了 解 更 多返回搜狐,查看更多 |


















【本文地址】