| MPOC 笔记 第十五章 (8)σ | 您所在的位置:网站首页 › σ键 › MPOC 笔记 第十五章 (8)σ |
MPOC 笔记 第十五章 (8)σ
|
一些补充:本节中出现了利用More O'Ferrall-Jencks,不代表周环反应中只有σ-迁移反应存在不同机理的竞争,只是前面的内容太多了。就像本书不停强调机理中的箭头不代表真实电子运动一样,机理间存在竞争,前面没强调只是因为本章讨论的是热周环反应。 比如[2+2]的周环加成都存在和双自由基等机理之间的竞争,之前举过异氰酸酯二聚的反应基本就不是周环反应,联烯和烯酮的二聚反应也不完全是环加成。看起来很像1,3-偶极子加成的Click反应一般认为机理如下[1]:  很多时候和初学者讲这么多混乱的东西会搅乱思路,所以基础有机教材里很少讲周环反应和其他机理的竞争。物理有机不是面向初学者的,很多问题未解决,很多混乱的地方也没厘清。 本节除了实践一下开头讲过的三种理论,还讨论了周环反应中的物理有机实验证据,最后补充了三个反应Cope重排、Claisen重排和ene反应。 15.5 σ-迁移重排 sigmatropic rearrangementσ 迁移是指σ 键在一个或多个π 体系上迁移的反应。用方括号中的数字来表明σ 键的移动,标示为[i, j]的σ 迁移重排是指侧面与一个或多个π 体系相接的σ 键迁移到一个新位置,离原来σ 键的位置距离分别为i 及j 。下面是最常见的两种σ迁移反应:[3, 3]-σ迁移和[1, 5]-σ迁移。类似电环化反应的对旋/顺旋,σ-迁移也有两种反应模式同面迁移或异面迁移,基团翻转或不翻转,所以所有的σ-迁移在特定的几何构型下都是允许的,要注意的不是允许禁阻,而是产物的立体化学。  [3, 3]-σ 迁移反应的特征结构比较明显,也常出现,其他σ-迁移反应比较少见一些,不过标记碳链看拓扑结构变化也能看出来。 这个反应和前面的电环化都可以用计算化学软件过渡态搜索试一试,网上有教程,因为做起来比较简单的,可惜放不了动图。  15.5.1 理论 15.5.1 理论下图是前线轨道理论(AB)、芳香过渡态理论(BC)和Woodward-Hoffmann规则(D)对σ迁移反应的分析。  基团进行[1, 3]-迁移时,要用σ 键的后部来保持同相位相互作用,这就使得迁移基团的立体化学反转(A图1)。而[1,5] 迁移则是迁移基团直接迁移到π 体系的末端,在过渡态中迁移基团仍是锥形的,因此迁移基团的立体化学是保持的(A图2)。要注意下面所画的过渡态轨道并不是这些结构的真实分子轨道,是简单构建出来的用以表示产生所观察到的立体化学的轨道相位分布。  σ-迁移的另一个问题是迁移基团迁移到这个π 体系的同面还是异/相反面,用前线分子轨道分析当然可以,用芳香性过渡态理论则更明快一些。见之前图的B和C。 闲话少说,只用芳香性过渡态理论来分析最经典的[3, 3]-σ迁移做一个实例,这个反应体系是没有节点的,是一个六电子的Hückel 体系,所以允许。  最后,可以用轨道对称性一般规则考察所有这些不同的迁移,又是翻转又是异面,有时容易错乱,极端的问题可能需要上Woodward-Hoffmann规则,之前有过极端的例题。  本章多次提到很多案例反应,比如丁二烯+乙烯的周环反应,其实不容易进行,很多时候要满足“电子需求”。然而, σ键迁移到环丙烷或从环丙烷环上迁移的反应却是一个极易发生的[3 , 3]-σ迁移反应,比如下面的例子。  更极端的例子是“瞬烯”,知乎上有人介绍这个结构,它具有如下所示三重对称结构(C3v 点群)。这个(CH)lO 结构极易经历一系列的[3,3]-σ 迁移,该过程的活化焓在11 kcal /mol 左右。在室温时,瞬烯的¹H 和¹³C NMR谱都只显示一个单峰,也就是说所有的碳原子以及所有氢原子都由于σ 迁移而相互等同。如果每个碳都独特标记的话,就会有10! / 3 = 1209600种相互转化的不同异构体。  15.5.2 实验观察:聚焦立体化学 15.5.2 实验观察:聚焦立体化学我们前面提到过, σ 迁移的两个重要因素是迁移基团的立体化学(翻转或保持)以及迁移发生在π 体系的同面和异面的立体化学问题。本章很少具体讨论实验问题,这一节大概是因为这个反应基础教材介绍有限吧。 如下图的[1, 5]氢迁移。在200 ℃下该反应通常表现出对迁移氢的一个较强的力学同位素效应(≈5),这表明了过渡态中发生了很大程度的键断裂,不记得可以回顾第八章。证明该反应确实经过了π 体系的同面发生,已由立体化学研究所证实:S/E 立体异构体(S 表示四面体碳原子的立体构型, E 表示双键的构型)。通过允许的同面氢迁移可以得到S/Z 和R/E 两种立体化学的产物,实际观察到了所预测的立体专一性,从而证实了重排的同面特性。读者应该可以自己理解受禁阻的反应情况(发生在π 体系的异面) ,它将会产生S/E 和R/Z 两种产物。  在1, 3- 环戊二烯中[1, 5] 氢迁移极易进行,此处环将π 体系锁定在一个利于氢迁移的理想几何形状上,该迁移过程的速率快到无法分离出单取代环戊二烯的各个单一的异构体,如方程式中所示的甲基环戊二烯,因为各异构体之间通过[1, 5]-氢迁移保持平衡,该反应看上去像是[1, 2]-迁移,如右上箭头转移所指,但它其实是一个六电子的过程,所以用[1.5]-迁移来描述更好。这个小故事经常出现在国外考试的小题目中,可以和碳正离子迁移的问题放一起讨论。  再看下面的反应,该反应对允许的[1, 5]-氢迁移非常容易进行,茚烯(indene) 在表现上的[1, 3]-σ 迁移实际上是两次[1, 5]-迁移的结果,在这里即使反应的第一步形成了高度不稳定的异茚(isoindene,不稳定是因为芳香性遭到破坏) 该迁移还是能够发生。  ps: 茚烯(indene)这个名字来自吲哚。 前面几个都是[1, 5]迁移,是因为底物都是环状平面结构的。[1, 3] 氢迁移必须在π 体系的异面发生,这是一个很难达成的几何结构。不过当迁移基团够大时,就可以达成了。碳向环状排列贡献一个sp³轨道,这样异面相互作用就变得可能了。如下图A ,迁移基团立体化学完全反转表明发生了异面相互作用。但如果是图B这种甲基取代的话,只有甲基可从碳骨架移开的反应物才可以发生迁移并导致立体化学完全反转 ,对于相反立体化学的反应物(图B下),在异面迁移过程中甲基必须向着碳骨架向上摆动。由于立体因素这不能发生,因此这个反应慢得多,并同时给出立体化学反转和保持的两种产物。  另一个很常见的σ 迁移是与碳正离子有关的[1, 2]-氢迁移,这个其实在第二章和第十一章介绍碳正离子的时候提过,虽然一般不会这么说,但这种迁移反应包含一个具有Hückel芳香性的两电子环状体系,从这种角度可以很好地解释为什么对碳负离子看不到类似的[1 , 2]迁移,因为相应的过程会涉及Hückel 反芳香性过渡态。   但负离子的形式上的[1, 2] 迁移可在含杂原子的体系中看到,比如Wittig 重排和Stevens 重排。根据上面的分析,它们大概率不属于周环反应,事实确实如此,Baldwin 在反应中使用筑标记的叶立德,表明迁移过程中立体构型保持。当反应在电子顺磁共振光谱仪中进行时,得到的证据表明至少在某种程度上反应中有自由基对形成。  上面这种杂原子参与的反应最常见的周环的是[2, 3]σ 迁移,这是一个六电子的过程,高级些的考试经常考。其中两个电子出自一对孤对电子,所以一共只含有五个原子。亚枫和各种类型的烯丙基叶立德都是常见的底物。必须注意的是,和简单的硫叶立德的[1, 2] Stevens 迁移不同,烯丙基硫叶立德的[2, 3]-迁移是一个协同的周环过程。[2, 3]-σ迁移己被证实在有机合成上有重要应用。在很多高等有机化学教程经常考察这个机理差异,类似的还有禁阻的[2+2]周环加成和自由基机理直接的竞争。  15.5.3 Cope 重排的机理 15.5.3 Cope 重排的机理很多时候基础教材看多了会形成一种错觉,比如觉得周环反应的机理不再有争议论,只要分析一下轨道对称性便可完全理解这些反应。但事实上,周环反应的很多基本方法和假设都曾遭到过质疑,并经受了大量实验和理论上的挑战。即使计算化学高速发展的今天还是有很多问题,很多之的发掘的地方,这也是为什么还在提出新方法的原因。本章的很多内容在本书出版后的几十年中有新的理解,要注意知识点的更新,尤其是周环加成。 有很多新的内容,比如16年的这篇nature:The pentadehydro-Diels–Alder reaction。  正因为周环反应研究最多的是环加成,Diels-Alder反应的机理研究历史很多教程中就有,所以本书下面举了另一个人名反应的例子,从历史发展的角度看看周环反应机理研究的历程——Cope 重排。 本章第一节介绍过,Diels-Alder反应发现很多年,相关理论才能从对称性的角度对这一类反应进行解释。同样的,A. Cope 在1940年发现了[3, 3]-σ 迁移重排反应,25年后关于这种反应轨道对称性守恒讨论的论文才被发表。  Cope重排:[3, 3]-σ 迁移 Cope重排:[3, 3]-σ 迁移Cope 重排的机理研究中有用的成果由Doering 和Roth 在1962 年报道,在这之前周环反应被认为“没有机理”,后来手性翻转方法在亲核取代反应机理中大放异彩,类似的方法便引入了Cope 反应的机理中。实验涉及利用3,4 -二甲基-1,5 -己二烯的两种立体异构体向2,6 -辛二烯的转化,为的是探究Cope 重排过渡态的几何形状。根据产物比率能看出椅型过渡态明显是优先的,或许这是因为椅型构象比船型构象更稳定。后期的一项研究用氘代替甲基作为立体化学标记得到了相同的结论,并且估计ΔΔG≠(船型-椅型)=5.8 kcal / mol。   关于Cope 重排反应究竟是不是一个协同的周环过程(下图A)是一个反复争论的议题,尤其是在多取代基的体系中,很容易就能想出包含双自由基的分步过程(下图B C)。此外,反应是否同步也曾是讨论的话题,我们可以想象三种极限反应途径:第一种A是同步的协同途径,形成一个对称的过渡态;第二种B是一个分步的机理,其中新σ 键在旧σ 键断裂之前形成;第三种C是在新σ 键形成之前,旧σ 键完全断裂。而我们知道,双自由基反应因为寿命短,立体选择性其实也很高。  对哪一种机理为主这个问题,最直接的评估方法就是热化学,1,5 - 己二烯的生成热为20. 2 kcal/mol , Cope 重排原型反应的活化能为34. 3 kcal/mol ,因此Cope 重排反应途径中的最高能量点的生成热近似为20. 2 kcal / mol +34. 3 kcal / mol = 54. 5 kcal /mol ( 用近似是因为Ea 和ΔH≠之间略有差异)。如果所提出的双自由基中间体的生成热显著高于该数值,我们就可以排除双自由基的机理。用第2 章中方法,可以估计出两个烯丙基自由基的生成热约为76 kcal/mol(上图C)。照这样,该双自由基中间体在Cope 重排原型反应中是不可能存在的,注意这个结论仅对原型反应成立,因为取代基可能可以稳定自由基。 对于环己烷双自由基(上图B)的情况。基团加和法估计生成焓在55 kcal/mol 左右,考虑到基因加和法对自由基的不确定性,并兼顾双自由基的情况,可见这个双自由基在Cope 重排中是有可能的。 取代基效应对于Cope 重排反应速率的影响也似乎与双自由基机理相吻合, 2 位和5 位的苯基以近似于加和的方式大大提高反应速率(4900 ≈ 69²) ,考虑到取代基处于重排中未经历成键或断键的碳上,这种效应尤其显著。为什么协同的周环反应会出现这样大的效应还不是很明确。因此,环己烷双自由基肯定是一种可能的中间体。  Gajewski 及其同事进行了一系列精巧的实验来探究Cope 重排反应的过渡态性质,所用的关键手段是二级动力学同位素效应。先来看看两个新定义:断键动力学同位素效应(BBKIE)和成键动力学同位素效应(BMKIE),这两者的比值被认为是一种用来表示过渡态中成键和断键发生的相对程度的很好量度。  当测定一系列己二烯衍生物的R 值时,发现了一些显著的变化。对于原型反应体系,有一个甲基取代时,R 值是1.8 ,表明在过渡态中成键略多于断键,但这么小的R 值当然不能支持一个像环己烷双自由基这样的过渡态。而在2 位和5 位的苯基取代,正如前面预期那样,显示了增加R 值的效应,致使在过渡态中键形成的程度明显超过键断裂的程度,因此过渡态中环己烷双自由基的特性有所增加,因为这个位置的苯基稳定了这样一种中间体。在3 位上连有两个强烈稳定自由基的氰基时,发现R 上面这段解释如果高等有机考解答题非常容易考。 很显然,过渡态的特性随着引人取代基的不同而变化,这使我们想起了More O'Ferrall-Jencks图。不过这个图的绘制方法和之前章节的略有不同。 为了通过一根轴评估反应过渡态中的成键程度,另一根轴估计断键程度,我们可以选用适当的动力学同位素效应与平衡同位素效应的比值。例如,成键轴表示整个反应中成键动力学同位素效应(BMKIE) 与热力学同位素效应之比,表示反应过渡态中键形成程度。断键轴的建立类似。  上图展示了过渡态结构随取代基的变化。左下角到右上角的对角线表示完全同步的理想参照态,其中表示成键百分数与断键百分数相等的过渡态位于中心点处。但一般所考察的结构并不位于这条线上,对于所有分析过的结构,在过搜态中成键和断键发生的程度都略有不同。但过渡态结构都位于从左上角到右下角的对角线上,这表明在过渡态中成键和断键的程度是紧密关联的,即成键百分数与断键百分数之和约等于100% 。这与本章多次展现的周环协同反应假想模型非常吻合。 最终这些实验研究仍不能完全排除Cope 重排中的双自由基中间体,对多取代的体系尤其如此,最后只有理论才可以对过渡态的结构给出一个明确的结论。在Cope 重排和其他周环反应过程中,理论和实验之间的互动有时是会引发争议的。此外,不同的理论模型时常会得出截然相反的预测,这进一步加剧了关于周环反应过渡态真实特性的争论。 近些年来,实验和理论都得出了相同的结论。因为现在可以用高水平的从头计算量子力学方法来探究周环反应的途径,这些研究始终且明确支持协同机理,对于Cope重排和其他诸如Diels - Alder 反应之类的原型周环反应,都有一个单一的芳香性过渡态。 目前用理论方法可以完全合理地解释在这些反应中所得到的大量的动力学同位素效应数据。例如,下面的四氘标记体系的成键和断键的动力学同位素效应的测量值分别为1.07±0.025 和0.89±0.018,而按协同芳香性过渡态的从头计算和DFT 计算结果则分别是1. 07 和0.88,对同一量值所做的精密实验测定和纯理论计算得到如此惊人的一致,为这类反应的协同周环过程提供了有力的证据。  书上有个补充问题:Oxy-Cope 重排中显著的取代基效应  这个问题考试遇到一般答阴离子取代基可以通过共振稳定自由基。  15.5.4 Claisen 重排 15.5.4 Claisen 重排在合成上最有用的σ 迁移反应是如式(1 5.42) 所示的Claisen 重排,该反应涉及烯丙基乙烯基醚向γ,δ-不饱和羰基化合物的转化,这是一个与Cope 重排密切相关的[3,3]-σ迁移反应。和Cope 重排不同的是Claisen 重排在本质上是不可逆的,这是因为羰基双键比烯烃双键稳定得多。  这个反应在生物反应中也存在。   Claisen 重排的一个重要变种是由Ireland 建立的烯醇酸醋(ester enolate) 的重排见下图。  机理研究 对于Claisen 重排的机理研究像Cope 重排一样,已经进行得十分广泛。动力学同位素效应再次起到了首要的作用,图A 列出了一些有代表性的kH/kD 数据。综合所有的证据表明Claisen 重排是一个协同周环机理,但并不奇怪该反应不是完全同步的。动力学同位素效应表明在过渡态中键的断裂显著多于键的形成,1. 09 >(1/ 0.976) ,More O'Ferrall-Jencks图偏左上角。对于烯醇酸醋Claisen 重排,两种同位素效应都更加显著一些,正常效应较大,逆同位素效应较小,,表明反应中断键仍旧比成键多,有硅氧基取代时过渡态沿此路径进行得更彻底。对于Oxy-Cope 重排,这种取代基效应可解释为由于硅氧取代基对氧代烯丙基自由基的稳定化作用。 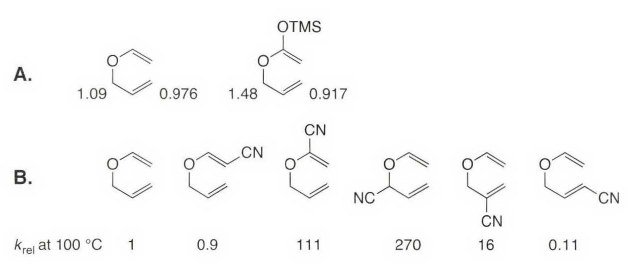 其他类型的取代效应也可能十分显著。 这种使周环加成反应不可逆,从而促进的方法。Diels-Alder反应做高分子时也会用到。  15.5.5 Ene 反应 15.5.5 Ene 反应Ene 反应是一类很重要的周环反应,但在某些方面很难归类。这类反应类似于一个σ 迁移反应,其中氢原子越过空间迁移到亲烯体(enophile ,氢受体)上。如果画出两种组分之间的虚键中两种反应物中的虚线J ,就可以将此反应看作[1,5]-迁移或者[3, 3]-迁移。不过这个反应国内教程更多的认为是环加成反应,这里烯丙基的C-H 键起到双烯中的第二根双键的作用。虽然归类方法有差异,但无论采取哪一种方式,都是一个六电子的周环过程。  Ene 反应的一个特别重要的类型是用单线态氧代替烯烃组分的反应,该反应的产物是烯丙基过氧化氢。   https://www.organic-chemistry.org/namedreactions/alder-ene-reaction.shtm https://www.organic-chemistry.org/namedreactions/alder-ene-reaction.shtm这个反应在一些高级的考试中很容易遇到,因为比较难想到。  https://www2.chemistry.msu.edu/faculty/reusch/virttxtjml/special0.htm https://www2.chemistry.msu.edu/faculty/reusch/virttxtjml/special0.htm1,写机理[2]:  分析在第十、十一章中多次讨论了联苯胺重排(Benzidine Rearrangement) 反应,这个反应早期被认为是多步完成的重排,后来经实验证实有部分是[5, 5]-sigmatropic,类似Claisen rearrangement。  因为Claisen rearrangement 比较经典,这个问题应该不难,所以核心问题是3-丁烯腈的π体系要怎么“连起来”。  “连”起来之后就比较容易了。  2,还是看几个真实物理有机习题集中的问题,APOC Problem 834。 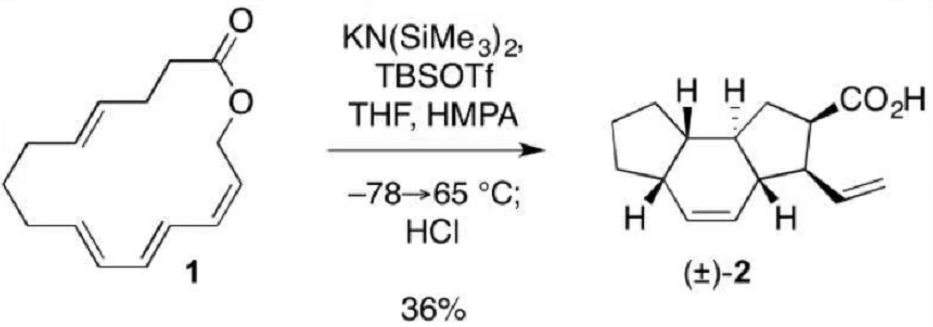 这是一种常见的考法:混入其他反应。这题明显要标号:  C4-C5 和C 9-12形成了一个环己烯的结构是Diels-Alder反应的特征产物,而C2-C13连接,C15-O1断开,是代表性的[3,3]-σ迁移反应,但是C1-C2之间没有双键,看反应条件可以想见是强碱是酯转换为了烯醇阴离子,本题就这么做出来了。本题另一个难点是立体化学,如果六元环画得多应该不难。  3,合成(题)中的妙用,σ-迁移可以带来夸张的碳链变化,下面两个例子出自《当代有机反应和合成操作》这本书,因为非常经典,很早就被出烂了。这种偷偷摸摸的σ-迁移非常恶心,但如果见识过应该就不会中招了。  4,还一种就是“硬考”,这个倒不多见。比如APOC problem 216,问产物的中新生成不对称中心的手性。  这题把σ迁移写在脸上,考的是对过渡态的理解,可以归入手性诱导问题。  5,APOC problem 919[3]  本题如果不是放在周环反应一章中,可能很多人都会觉得莫名其妙,这就是ene反应的难点,想不到。本题还是一道逆ene反应(retro-ene reaction)。  上一节: 下一节: 目录: 《现代物理有机化学》笔记 第一章 化学键基础(1)《现代物理有机化学》笔记 第二章 张力和稳定性(1)《现代物理有机化学》笔记 第三章 溶液和非共价结合力(1)《现代物理有机化学》笔记 第五章 酸碱(1)《现代物理有机化学》笔记 第四章 分子识别和超分子化学(1)《现代物理有机化学》笔记 第六章 立体化学(1)《现代物理有机化学》笔记 第七章 动力学(1)《现代物理有机化学》笔记 第八章 机理相关实验(1)《现代物理有机化学》笔记 第九章 催化(1)《现代物理有机化学》笔记 第十章 机理(1)《现代物理有机化学》笔记 第十一章 机理2(1)《现代物理有机化学》笔记 第十二章 金属有机(1)《现代物理有机化学》笔记 第十三章 有机聚合物和材料化学(1)《现代物理有机化学》笔记 第十四章 电子结构理论新概念(1)《现代物理有机化学》笔记 第十五章 热周环反应(1)《现代物理有机化学》笔记 第十六章 光化学(1)《现代物理有机化学》笔记 第十七章 有机电子材料(1)《现代物理有机化学》笔记 附录五参考^Direct Evidence of a Dinuclear Copper Intermediate in Cu(I)-Catalyzed Azide-Alkyne Cycloadditions https://science.sciencemag.org/content/340/6131/457^https://onlinelibrary.wiley.com/doi/abs/10.1002/anie.201900434^https://doi.org/10.1021/ja0670660 |
【本文地址】