| 聚合酶链反应 PCR | 您所在的位置:网站首页 › pcr缓冲液中镁离子的作用是 › 聚合酶链反应 PCR |
聚合酶链反应 PCR
|
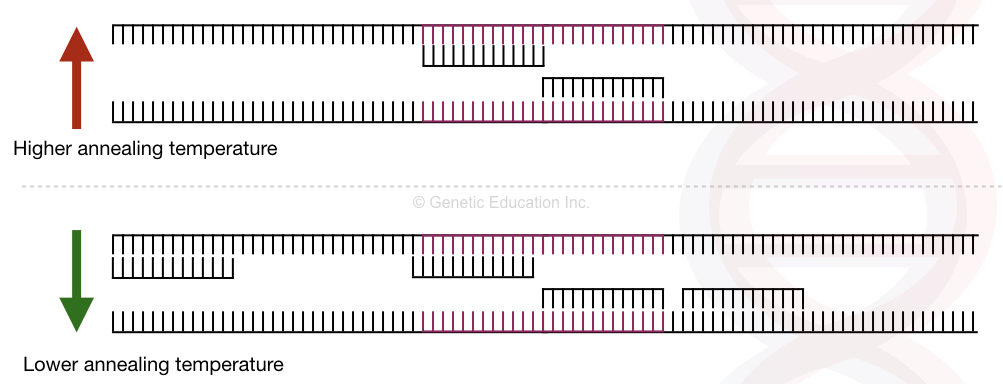
聚合酶链反应(Polymerase Chain Reaction, PCR)是 Kary Mullis 在 20 世纪 80 年代开发的用以快速扩增特定 DNA 片段的革命性方法。PCR 的基础是利用 DNA 聚合酶合成与所提供的模板链互补的新 DNA 链。因为 DNA 聚合酶只能将核苷酸添加到先前存在的 3'-OH 基团上,所以 PCR 反应需要一个带有3'-OH 基团的引物。引物的使用,同时也让有针对性地扩增特定区段成为了可能。在 PCR 反应结束时,特定序列将累积成数十亿个拷贝(扩增子 amplicons)。 目录1 PCR 原理1.1 表示 PCR 反应的两种方法2 PCR 反应的组分2.1 DNA 模板2.2 引物2.2.1 设计引物2.2.2 计算退火温度2.2.3 引物的纯化方法2.3 DNA 聚合酶2.3.1 Taq DNA 聚合酶2.4 dNTPs2.4.1 dNTPs 添加量2.4.2 dNTPs 的使用与储存2.5 缓冲液2.5.1 二价离子缓冲液2.5.2 KCl 缓冲液2.5.3 pH 缓冲液2.6 特殊添加剂3 PCR 反应的类型3.1 基本 PCR 反应3.1.1 反应体系3.1.2 阳性对照和阴性对照3.1.3 反应结束3.1.4 验证 PCR 扩增产物3.1.5 纯化 PCR 扩增产物3.1.6 其他3.2 Inverse PCR3.3 PCR Amplification of GC-Rich Templates3.4 Long and Accurate PCR (LA PCR)3.4.1 TENB Buffer3.4.2 其他3.5 Nested PCR3.6 Screening Colonies by PCR3.7 Hot Start PCR3.7.1 一种热启动 PCR 的实验方案3.8 Touchdown PCR (TD-PCR)3.9 Multiplex PCR3.9.1 多重 PCR 的引物设计3.9.2 多重 PCR 的优势3.9.3 Relay-PCR3.10 Reverse Transcription PCR (RT-PCR)3.10.1 RT-PCR 的引物3.10.2 RT-PCR 使用的反转录酶3.10.2.1 AMV 和 MuLV3.10.2.2 rTth DNA 聚合酶3.10.3 RT-PCR 步骤3.10.4 One-step & Two-step RT-PCR3.10.5 RT-PCR 中的对照3.10.6 Ung/dUT 系统3.11 RACE3.11.1 3' RACE3.11.2 5' RACE3.12 Real-time Quantitative PCR (real-time qPCR)3.12.1 qRT-PCR 的步骤3.12.2 qRT-PCR 的内参 1 PCR 原理PCR 利用温度循环启动和结束酶催化 DNA 的合成,包含 3 个步骤: 热变性 Denaturing:模板 DNA 的热变性,通常温度 >90 ℃. 在 94°C–95°C 变性 30 个循环对 DNA 聚合酶的酶活性的影响是可以不计的。 在 PCR 的第一个循环中,通常增加 5 min 的变性时间,以便长链 DNA 分子充分变性。 对于 G + C 含量为 55% 或更低的线性 DNA 模板的常规扩增,建议在 94°C–95°C 下变性 45 秒。对于更高 G+C 含量的模板,推荐使用从古菌中分离的 DNA 聚合酶(热稳定性更好),如从 Pyrococcus furiosus 中分离的 Pfu 聚合酶。 退火 Annealing:将 2 个人工合成的 寡核苷酸引物 (oligonucleotide primers) 退火至预先变性的模板 DNA。 退火温度 Ta 通常比寡核苷酸引物与其模板解离的计算解链温度 Tm 低 3–5°C。 优化退火温度通常从 Tm-2 开始,Tm-10 截止,中间设置梯度进行测试。 延伸 Extension:其中DNA合成起始于结合引物的 3′ 端。在耐热 DNA 聚合酶催化的酶促反应中,引物的延伸发生在大约 55-70 ℃ 之间的温度。一般情况下,1 min 能延伸 1000 bp。PCR 在热循环仪中进行。最初的两个循环,产生长度与目标片段 DNA (两引物之间,包含两引物)相等的扩增片段。之后的每个循环,目标片段的数量均加倍,目标片段数量呈指数形式增长,直至引物或寡核苷酸缺乏。
[!Ct] Ct 值被定义为在基线上方首次检测到荧光信号的循环次数,与目标序列初始拷贝数的 log10 负相关。在大多数商用机器中,Ct 值被设定为信号比平均基线信号高 10 倍时的循环数。 在 PCR 反应因为原料缺失或产物抑制等原因达到终点时,在两种表示方法上都会形成一条与 \(x\) 轴平行的线。在 产物量(荧光强度)-循环数图中,不同的反应终点有显著的不同。但在 log10 产物量-循环数图中,不同反应最终达到同一个平台,因为用对数表示的情况下产物量差距不大。 2 PCR 反应的组分 2.1 DNA 模板 DNA 模板:包含目标序列的样本 DNA。在反应开始时,对原始双链 DNA 分子施加高温以使双链彼此分离。 线性单双链 DNA 比闭合环状 DNA 的扩增效率高 2.2 引物 引物:与目标序列互补的单链DNA短片段。聚合酶从引物末端开始合成新的DNA。 一对或几对 引物浓度:一般每个引物的浓度在 0.1-0.5 μM 高或低(此浓度通常足够 1-kb 的 DNA 片段扩增 30 个循环) 引物浓度高:错配,非特异性扩增 引物浓度低:产物浓度不够[!Note] 合成的引物 5' 没有磷酸基团。 2.2.1 设计引物引物设计步骤: 分析目标基因的潜在引物位点,这些引物位点需不含同聚体片段,没有形成二级结构的明显趋势,不具有自互补性,并且与目标基因组任一链上的其他序列都没有显著的同源性。 根据引物设计原则列出可能的正向引物和反向引物,并计算出熔解温度(Tm)。 选择 G+C 含量相似且在 40%~60% 之间,并能扩增出大小和碱基组成适当扩增产物的引物对。 优化引物的长度和/或位置,使 3′ 末端核苷酸为 G 或 C。检查两个引物是否显示出明显的互补性。根据经验,一个引物上与另一个引物互补的核苷酸不得超过三个。引物设计原则: 碱基组成:G + C 含量应在 40% 和 60% 之间,所有四个碱基沿引物长度均匀分布(例如,没有多聚嘌呤或多嘧啶束,也没有二核苷酸重复)。如果可能,避免容易形成二级结构的富含 GC 的延伸。 长度:单个引物长度 18-30 bp,常用 21 bp 左右。 5' 外切酶活性(不具备校对活性):类似 Klenow 片段中的校对活性区段失活。在 74˚C 下,一个酶活力单位 U 的 Taq DNA 聚合酶在 30 分钟内将 10 nmol 的 dNTP 掺入。尽管 Taq 聚合酶仍然是常规扩增 DNA 小片段的首选酶,但它缺乏 3'->5' 校对功能,并且错误掺入率高(9000 个核苷酸中的一个核苷酸)。从其他嗜热细菌和古细菌中分离出来的耐热 DNA 聚合酶,例如 Pfu DNA 聚合酶,具有校对活性。可以在需要更高保真度时代替 Taq(或与 Taq 结合使用),当目标扩增子的长度超过几千个碱基,或者在通过逆转录 PCR (RT-PCR) 克隆 mRNA 时。 2.4 dNTPs核苷酸(dNTPs 或脱氧核苷酸三磷酸):碱基A、T、G和C的单个单元,是新DNA链的“构建块”。标准 PCR 包含等摩尔量的 dATP、dTTP、dCTP 和 dGTP。 2.4.1 dNTPs 添加量核苷酸加入的量需根据具体反应条件来确定。例如,在使用 Taq 酶,1.5 mM 的 \(MgCl_2\) 条件下,每一种 dNTP 的含量在 200-250 μM。 高浓度的 dNTP (>4 mM) 能够抑制 PCR 反应,可能是由于 dNTP 的磷酸基团螯合 \(Mg^{2+}\) 使其浓度下降的原因。
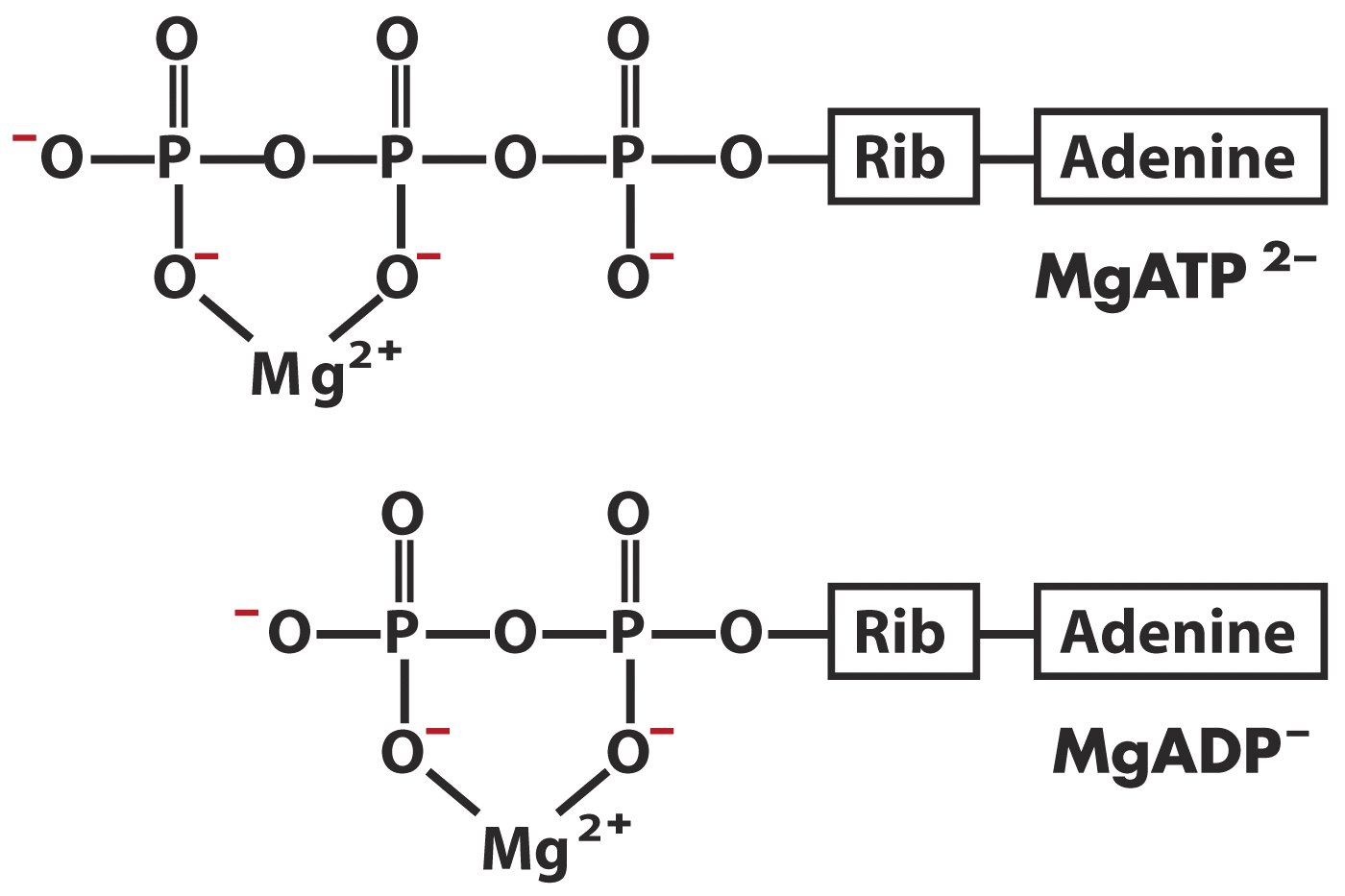
dNTPs 储液对冻融敏感,经过几次冻融循环后,PCR 的效率开始下降。为避免此情况出现,应将 100-200 mM 的 dNTPs 储液分成 2-5 μL 的小份(含 10 mM Tris pH 8.0),并在两次冻融后丢弃。如果不加入缓冲液,可能会使 dNTPs 发生酸水解。 2.5 缓冲液 2.5.1 二价离子缓冲液DNA 聚合酶发挥作用需要二价金属离子的辅助。 2 个二价的金属阳离子,结合在 DNA 聚合酶由 β 折叠片构成的手掌域催化位点附近,改变正确碱基配对的 dNTP 和引物 3'-OH 周围的化学环境。第一个金属离子降低 3'-OH 对氢的亲和力,产生一个准备对引入 dNTP 上 α-磷酸进行亲和攻击的 3'-\(O^-\). 第二个金属离子与 dNTP 的 β- 和 γ-磷酸负电荷协同作用,稳定由连接反应之后产生的焦磷酸。 PCR 反应中常用的二价离子为镁 \(Mg^{2+}\) 离子,有些缓冲液也使用锰 \(Mn^{2+}\) 离子,钙 \(Ca^{2+}\) 离子作用效率最低。 镁离子在 PCR 反应中的作用 与 dNTP 形成复合物,作为 Taq 酶的底物 稳定引物-模板复合物镁离子的用量:超出引物+模板的磷酸基团量(一般采用 1.5 mM) \(Mg^{2+}\) 过多,会导致 DNA 双链结合地过于稳定,导致热变性不彻底。 \(Mg^{2+}\) 过少,引物与模板配对的特异性降低,增加镁离子的量有助于减少错误配对(4.5 mM - 6 mM)PCR 反应体系中不应存在螯合剂,如乙二胺四乙酸、\(PO_4^{3-}\),以使 \(Mg^{2+}\) 正常发挥作用。 [!Caution] 反复冻融氯化镁溶液会使之分层,使用前需完全混匀。 2.5.2 KCl 缓冲液KCl 在 PCR 中主要起到中和 DNA 骨架上负电荷的作用。能增加引物与模板配对的稳定性。 标准 PCR 缓冲液含有 50 mM KCl,适用于扩增长度大于 500 bp 的 DNA 片段。将 KCl 浓度提高到 70-100 mM 通常会提高较短 DNA 片段的产量。 2.5.3 pH 缓冲液Tris-HCl 目的是让 72 摄氏度附近的 pH ~7.2。所以加入室温 pH 8.3-8.8 的缓冲液 2.6 特殊添加剂在调整 \(Mg^{2+}\) 、\(K^+\) 浓度仍然不能提高 PCR 效率或特异性时,可以考虑以下添加剂。 减少二级结构 二甲基亚砜 Dimethyl Sulfoxide, DMSO:助溶剂,在胞嘧啶残基处与 DNA 结合并改变其构象,从而使 DNA 对热变性更加不稳定,同时减少模板或引物中的二级结构。对 GC 丰富的模板特别有用。使用浓度:5-10% 非离子和阳离子去污剂 Triton X-100, Tween 20, NP-40:降低 DNA 二级结构,或减弱 SDS 的负面影响。使用浓度:0.25-1% 甜菜碱 Betaine:使用甜菜碱一水合物(Betaine monohydrate)可提高 DNA 小沟中富含鸟嘌呤和胞嘧啶区域的鸟嘌呤和胞嘧啶的水合作用,影响DNA分子结构,减少 DNA 中的二级结构。使用浓度:1-3 M 甘油 Glycerol:减少二级结构。使用浓度:5-10% 提高特异性 甲酰胺 Formamide:和DNA中的大沟和小沟相结合,从而降低母版 DNA 双螺旋的稳定性,降低解链温度 Tm。增加引物退火的严格性,从而减少非特异性引物并提高扩增效率。使用浓度:1-10% 四甲基氯化铵 Tetramethylammonium Chloride, TMAC:增加配对的特异性,提高解链温度 Tm,减少可能存在的 DNA-RNA 配对。使用浓度:10-100 mM 牛血清白蛋白 BSA:对于可能被腐殖酸污染的模板(例如被土壤污染的环境样品)非常有用,还可以防止反应组分粘在管壁上。使用浓度:0.8 mg/ml 谷氨酸钾 potassium glutamate, KGM:PCR 反应中的酶在细菌或古菌之外发挥作用,采用细菌中大量存在的谷氨酸盐(KGM)代替 KCl 能模拟细菌内部环境,使 DNA 聚合酶更好发挥作用。谷氨酸盐在体外能显著增加 蛋白质-DNA 互作。可能降低核苷酸的错误掺入率,并对长模板的扩增有帮助。 硫酸铵 ammonium sulfate:\((NH_4)_2SO_4\) 通过破坏错配过程中出现的弱氢键来帮助防止引物和模板 DNA 之间的错配,并增加 \(KCl\) 和 \(MgCl_2\) 的浓度宽容范围。 7-deaza-2′-deoxyguanosine: 一种 dGTP 类似物,对于极富 GC 的模板(有 83% GC 的报道)特别有用。使用浓度:\(dGTP:7-deaza-2′-deoxyguanosine = 1:3\) 3 PCR 反应的类型 3.1 基本 PCR 反应基本聚合酶链式反应 (PCR) 已适用于分子克隆中的各种任务,包括 DNA 测序、体外诱变、突变检测、克隆 cDNA 和基因组 DNA 以及等位基因。 3.1.1 反应体系 Component Volume Amplification Buffer (10x) 5 μL dNTP mix (20 mM each, pH 8.0) 1 μL Forward primer (20 μM) 2.5 μL Reverse primer (20 μM) 2.5 μL Thermostable DNA polymerase (1–5 units/µL) 1-2 units Template DNA 5-10 μL Water (PCR grade) to a final volume of 50 μL 3.1.2 阳性对照和阴性对照阳性对照用来监测 PCR 的效率,阴性对照用来检测含有目标序列的 DNA 的污染。 ByStander DNA Template DNA Target DNA Specific primers Positive control 1 + - + + Positive control 2 - - + + Negative control 1 - - - + Negative control 2 + - - -目标 DNA(Target DNA):包含目标序列。它可以是重组 DNA 克隆、纯化的 DNA 片段或基因组 DNA 样本。它应该以与模板 DNA 中预期的浓度相当的浓度添加到阳性对照中。通常需要准备一系列包含不同数量的目标 DNA 的阳性对照,浓度跨越模板 DNA 中预测的数量。 3.1.3 反应结束 一个成功的 PCR 反应应该会产生一个易于看到的预期大小的 DNA 片段。条带正确与否可以通过 DNA 测序、Southern 杂交、或限制性作图来进一步确认。 含有两个阳性对照样品和待测模板 DNA 的凝胶泳道应包含一条具有适当分子量的 DNA 条带。包含阴性对照样本的泳道中应该不存在该条带。 3.1.4 验证 PCR 扩增产物当通过琼脂糖凝胶电泳分析产物时,理想情况下应仅存在 1 条带,若多条带同时存在,则采用如下方法: 方法 1:采用 Hot Start PCR 或 Touchdown PCR 重复实验。若不成功,提高退火温度继续尝试。 方法 2:将不同的 DNA 条带分别回收,作为模板进行 PCR,如果可能,采用嵌套引物。如果仍出现多条条带,对最大的条带进行测序。若上述两种方法均不奏效,将所有条带从胶上回收,采用 TA 克隆分别进行克隆。 3.1.5 纯化 PCR 扩增产物纯化 PCR 产物主要是除去体系中的 dNTP、单链 DNA、引物、DNA 聚合酶。除了使用酚氯仿法 PCl 等传统方法,纯化 DNA 的试剂盒有几种类型: DNA 与硅胶表面结合,洗涤,然后洗脱。 DNA 与具有阴离子交换能力的磁珠结合。 DNA 通过装有弱阴离子二氧化硅的微量移液器吸头中的离心柱纯化。 3.1.6 其他 当 PCR 仪没有热盖(hot lid)时,以 50 μL 的轻质矿物油覆盖反应体系。反应结束之后,可以用 150 μL 氯仿对产物进行抽提。含有水的 DNA 会在甘油与氯仿之间形成胶束,采用微量移液器吸取此部分。 PCR 产物可在 4 ℃ 过夜,之后需转移至 -20 ℃ 的冰箱内。 3.2 Inverse PCR标准聚合酶链式反应 (PCR) 用于扩增位于两个内向引物之间的 DNA 片段。相比之下,反向 PCR Inverse PCR(也称为 inverted or inside-out PCR) 用于扩增位于已知 DNA 序列一端没有可用引物的未知 DNA 序列。
反向 PCR 广泛用于快速等位基因分析和确定逆转录病毒、转基因和转座子集成到基因组中的位置。它是一个耗时且繁琐的过程。与常规 PCR 相比,限制性消化和连接测定需要更多时间。该技术依赖于如此多的酶促步骤,因此反应失败的可能性很高。整个实验的成本高于传统的PCR。 3.3 PCR Amplification of GC-Rich Templates在哺乳动物的基因组中,通常存在着许多具有特征的序列。这些特征包括:单核苷酸卫星序列、反向重复序列、GC-富集的区段。在基因组 PCR 时,这些具有特征的序列往往会形成二级结构,不能很好地解链。同时,被阻碍的 DNA 聚合酶会产生许多短的、不正确的扩增产物,这些产物在之后的循环中也会被放大。这些问题最终都会导致 PCR 失败。 有时候,更改引物设计,更改循环方案,或使用 Hot Start PCR、 Touchdown PCR 的组合能够解决问题。更常见的,是使用多管齐下的方法,例如在扩增反应中使用增强剂、调整循环方案,以及在必要时设计新的引物组。 引物设计 在设计用于扩增富含 GC 的序列的引物时,使用寡核苷酸设计程序检查: 引物之间的二聚体形成及其在靶标上的结合位点结合的吉布斯自由能 (ΔG) 二级结构的吉布斯自由能 (ΔG)添加剂 大多数情况下,多种添加剂混合使用的成功率比单一添加剂高。 Additive Solution (5x) Reagent Quantity (for 10 mL) Final Concentation Betaine (5 M) 5.4 mL 2.7 M Dithiothreitol (1 M) 67 µL 6.7 mM Dimethyl sulfoxide 670 µL 6.7% (v/v) Bovine serum albumin (2 mg/mL) 275 µL 55 µg/mL循环条件 将升温 (2.5 ˚C/sec) 和降温 (1.5 ˚C/sec) 速度减慢是应对 GC-含量高的目标序列的一种方法。 3.4 Long and Accurate PCR (LA PCR)标准 PCR 反应对小于 ~3kb 的目标 DNA 序列的扩增十分有效,但对于更长的区段,则会产生不同长度的截短产物,在凝胶上显示出涂抹 smear 现象。Long and Accurate PCR (LA PCR) 采用两种不同 DNA 聚合酶的混合来进行 PCR 反应。 第一种 DNA 聚合酶(混合中占比高)高效但易错 第二种 DNA 聚合酶(混合中占比低)具有校对活性(3'->5' 核酸外切酶活性)商用 DNA 聚合酶产品: Expand Long Template PCR System [Roche] Advantage HF 2 PCR Kit [Clontech] KlenTaq LA [DNA Polymerase Technology] 3.4.1 TENB BufferLA PCR 中的模板 DNA 最好溶于 TENB buffer。 Reagent Quantity (for 10 mL) Final Concentation Bovine serum albumin (2 mg/mL) 0.5 mL 100 μg/mL EDTA (0.5 M) 2 μL 0.1 mM NaCl (1 M) 100 μL 10 mM Tris-Cl (1 M, pH 7.9) 100 μL 10 mM 3.4.2 其他长片段 DNA 扩增失败的原因: 模板或产物 DNA 在高温时损坏(缓冲液无法在高温时控制 pH)。 存在可以促进 DNA 在高温下裂解的杂散二价阳离子(应考虑 \(Mn^{2+}\))。 变性温度或时间不恰当,长 DNA 分子不容易解链。 热稳定性聚合酶(如缺乏编辑功能的 Taq)导致错误碱基高频掺入,在 3' 插入不匹配的碱基从而导致聚合酶停滞。对 LA PCR 的进一步改进: 加入热稳定的尿苷三磷酸酶 UTPase。在 PCR 中,dCTP 的水解会产生 dUTP,在引物中的 dUTP 会使古菌的 DNA 聚合酶 Pfu 与之紧密结合并被抑制。 加入高浓度的甜菜碱,终浓度 1.3 M。能够让 LA PCR 使用较低的变性温度(from 94˚C to 91˚C–93˚C)。 将目标 DNA 的延伸时间增加到 2 min/kb。 3.5 Nested PCR嵌套聚合酶链式反应 Nested PCR 用于需要增加 PCR 的敏感性和/或特异性的情况,例如: 扩增多态基因家族的特定成员。 扩增基因的 cDNA 拷贝时(当 mRNA 在含有异质细胞群的临床标本中以非常低的丰度存在时)。嵌套 PCR 通常涉及两个连续的扩增反应,每个反应使用不同的引物对。第一次扩增反应的产物用作第二次 PCR 的模板,第二次 PCR 由位于第一引物对内部的寡核苷酸引发。使用两对寡核苷酸可以进行更多的循环,从而提高 PCR 的灵敏度。反应特异性的提升,源于使用两组不同的引物与同一目标模板的结合。嵌套 PCR 也是一种,在了解目标序列的前提下,扩增长模板片段的有效方法。
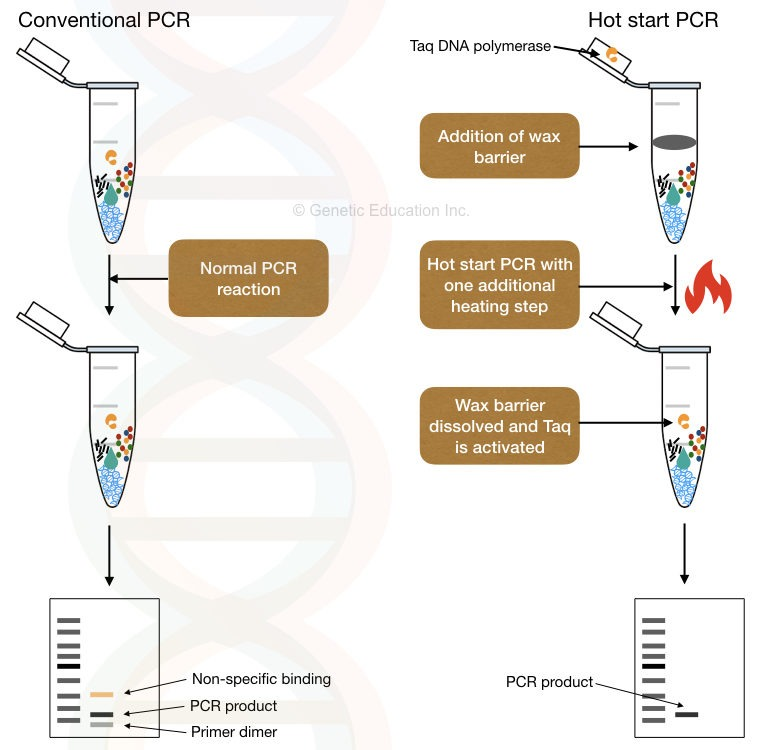
在嵌套 PCR 中,引物分别叫做 Outer Oligonucleotide 和 Inner Oligonucleotide. 还有一种半嵌套 PCR(semi-nested PCR) 其中的一条 Inner Oligonucleotide 与一条 Outer Oligonucleotide 相同。半嵌套 PCR 的特异性没有嵌套 PCR 高。 3.6 Screening Colonies by PCR菌落 PCR 是一种用于在转化步骤后快速筛选在选择性培养基上生长的酵母菌或细菌菌落的方法,以验证所需的基因构建体是否存在,或扩增构建体的一部分。聚合酶链式反应 (PCR),而不是更费力的小量制备,通常用于筛选大肠杆菌菌落中包含感兴趣序列的重组质粒。当筛选通过 PCR 扩增的 DNA 片段的菌落时,可以使用与扩增反应相同的引物进行筛选。为了探明克隆片段的方向,可以使用第三个引物,它与紧邻插入位点的质粒序列结合并指向插入物。 3.7 Hot Start PCR由于 PCR 反应体系在室温下配制,容易得到非特异性的扩增产物,热启动 PCR Hot Start Polymerase Chain Reaction 是一种减少此种产物的常用技术。在室温下,PCR 引物可以与不完全互补的模板序列退火。由于热稳定的 DNA 聚合酶在这些低温下具有部分活性,因此聚合酶可以延伸错误退火的引物。然后这个新合成的非特异性区域作为模板,延伸和合成非特异性的扩增产物。 热启动 PCR Hot Start Polymerase Chain Reaction 还可以减少引物二聚体的形成。在较低温度下,PCR 引物可以通过互补区域相互退火,DNA 聚合酶可以延伸退火的引物以产生引物二聚体,在溴化乙锭染色的凝胶上通常显示为大约 50-100 bp 的扩散带。
为了使 PCR 反应在加热到特定温度之前不会进行,通常有几种方法: 在达到延伸温度前的体系中省略掉 1 个或几个关键组分,达到温度后再加入。 将 1 个或几个关键组分物理隔离。例如可以将 \(Mg^{2+}\) 或 DNA 聚合酶包埋在蜡珠(wax bead)中,蜡珠会在变性温度下融化,释放组分。 通过结合其他分子,将 DNA 聚合酶在达到特定温度前可逆失活。这些分子在加热之后会分离或失活。 采用适体 aptamer(适体通常是一段寡核苷酸) 采用抗体 通过化学修饰使 DNA 聚合酶在常温下失活。 采用修饰过的引物,在空间上阻止 DNA 聚合酶在低温下掺入核苷酸。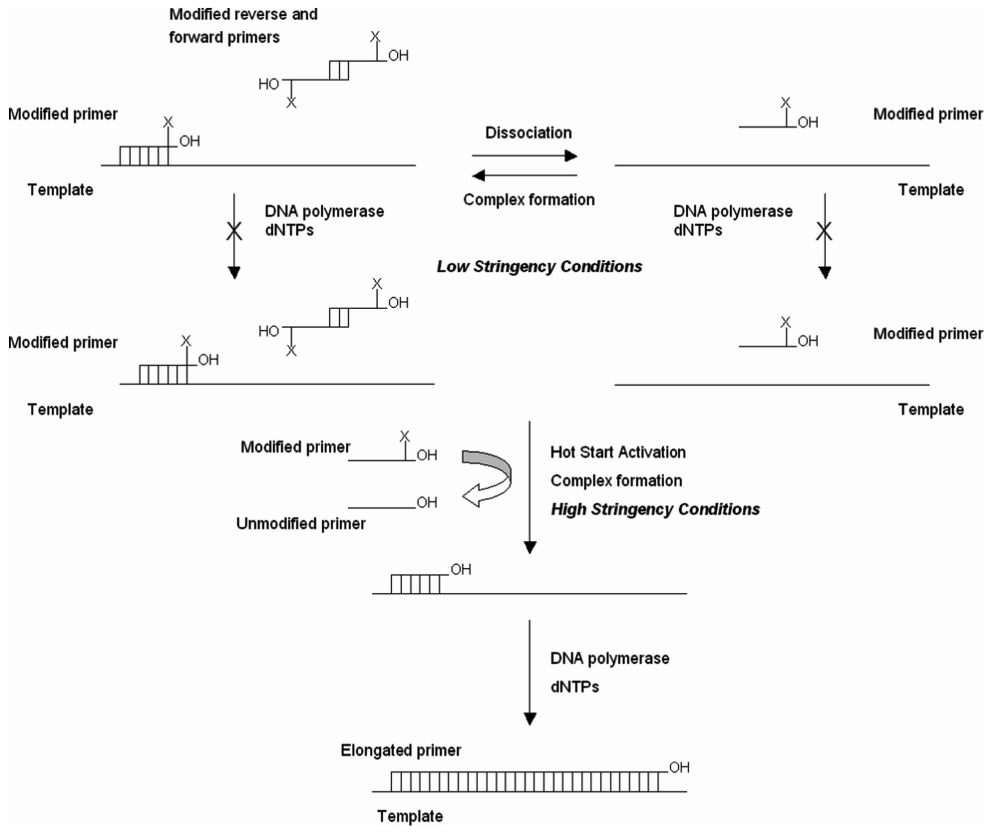 3.7.1 一种热启动 PCR 的实验方案
配制下层试剂混合物:
Component
Volume
Amplification Buffer (10x)
5 μL
dNTP mix (20 mM each, pH 8.0)
1 μL
Forward primer (20 μM)
2.5 μL
Reverse primer (20 μM)
2.5 μL
Water (PCR grade)
to a final volume of 46 μL
将 46 μL 的下层试剂混合物放入 PCR 管中,之后放入蜡珠。
关闭 PCR 管,放入 80°C 的热循环仪中 5 min。
从循环仪中取出 PCR 管。在 20°C 下放置 5-10 min。
同时,配制上层试剂混合物:对于每个反应,将 1–2 单位的 Taq DNA 聚合酶与 5 μL 的模板 DNA 混合。
将 PCR 管放入热循环仪中:
Cycle Number
Denaturation
Annealing
Polymerization
1
1 min at 94˚C
2-31
45 sec at 94˚C
30 sec at 55˚C
1 min at 72˚C
Last Cycle
7 min at 72˚C
3.8 Touchdown PCR (TD-PCR)
3.7.1 一种热启动 PCR 的实验方案
配制下层试剂混合物:
Component
Volume
Amplification Buffer (10x)
5 μL
dNTP mix (20 mM each, pH 8.0)
1 μL
Forward primer (20 μM)
2.5 μL
Reverse primer (20 μM)
2.5 μL
Water (PCR grade)
to a final volume of 46 μL
将 46 μL 的下层试剂混合物放入 PCR 管中,之后放入蜡珠。
关闭 PCR 管,放入 80°C 的热循环仪中 5 min。
从循环仪中取出 PCR 管。在 20°C 下放置 5-10 min。
同时,配制上层试剂混合物:对于每个反应,将 1–2 单位的 Taq DNA 聚合酶与 5 μL 的模板 DNA 混合。
将 PCR 管放入热循环仪中:
Cycle Number
Denaturation
Annealing
Polymerization
1
1 min at 94˚C
2-31
45 sec at 94˚C
30 sec at 55˚C
1 min at 72˚C
Last Cycle
7 min at 72˚C
3.8 Touchdown PCR (TD-PCR)
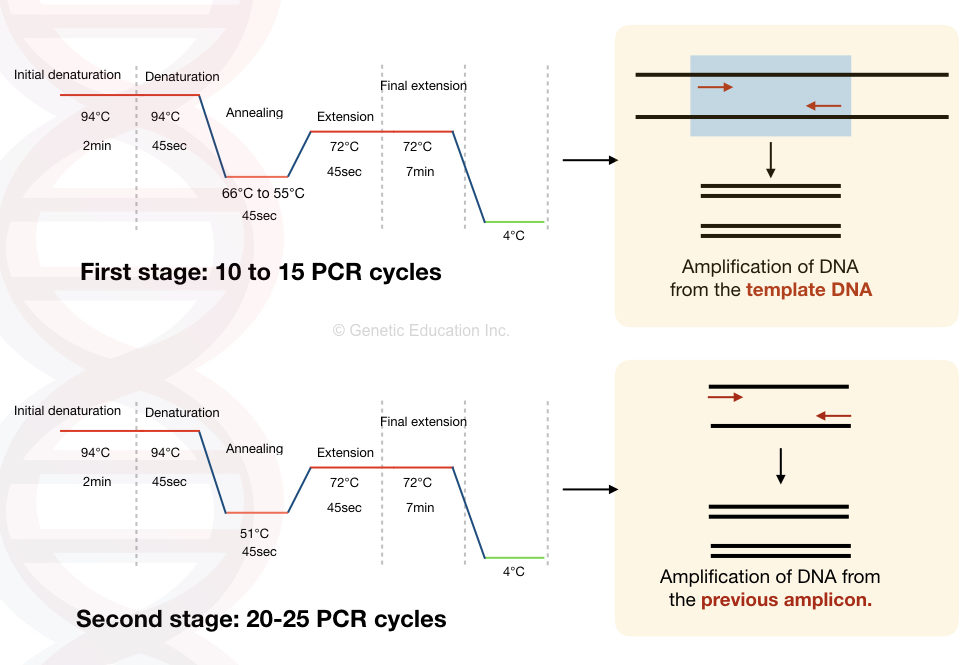
对于许多研究人员来说,降落 PCR,touchdown PCR 绕过了为每对引物确定最佳退火温度的需要,并用于在常规 PCR 中获得可接受的扩增产物产量。当目标序列与引物序列可能不匹配时(例如从氨基酸序列推导引物序列),降落 PCR 是绝佳的选择。 在 PCR 反应中,非特异性的配对能导致目标序列之外的序列被扩增,在温度低的时候尤为明显。产生的错误扩增产物在之后的循环中被指数放大。
在 touchdown PCR 中,选择的退火温度比解链温度 Tm 高 5-10 °C。因此,引物只能与它们的特异性和互补序列结合。每次循环后,温度降低 1 °C,最终达到低于解链温度 2-5 ℃。然后,PCR 循环的退火温度在最低退火温度保持 12-27 个循环。即使由于较高的温度,目标 DNA 区段在初始循环中没有被扩增,它也会在后续循环中被扩增。在较高温度获得的正确目标片段在之后的循环中被大量扩增,由于序列的富集呈指数形式,所以正确的片段在产物中占绝大多数。
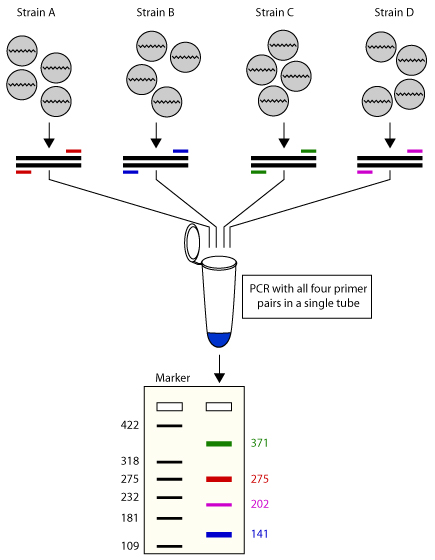
多重 PCR (Multiplex PCR) 用于在单个 PCR 实验中扩增多个靶标,可以通过在反应混合物中使用多个引物对来扩增多个目标序列。其最初被用来筛选人类基因组 Duchenne 肌营养不良基因座的不同突变。作为 PCR 实际应用的延伸,该技术有可能在不影响实验效用的情况下,在实验室内节省大量时间和精力。 多重 PCR 有两种类型: 单模板 PCR:此技术采用单个模板(例如基因组 DNA),采用多个引物对扩增模板中的多个特定区域。 多模板 PCR:采用多个模板与引物对。多个引物的存在可能导致彼此交叉杂交以及与其他模板错误配对。
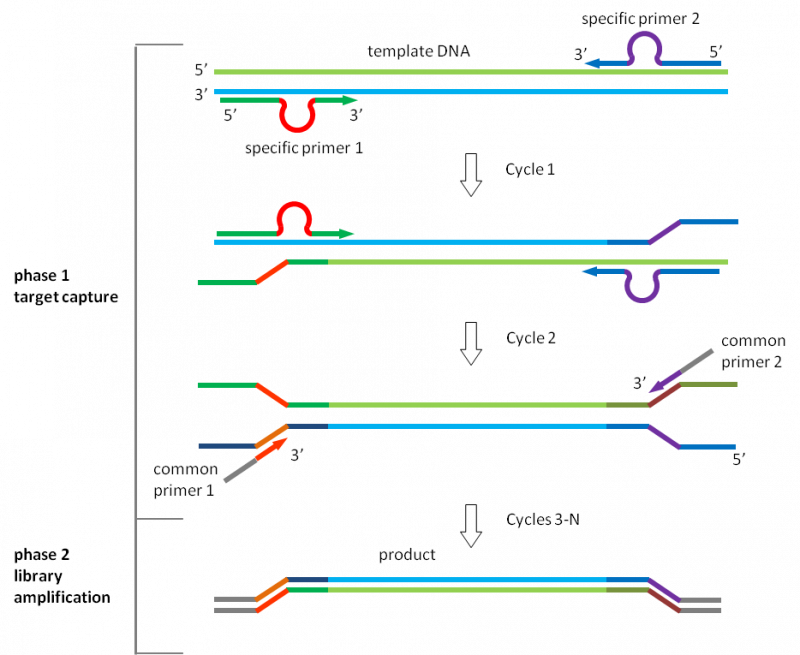
多重 PCR 的引物设计有如下特点: 引物长度:通常采用长度较短的引物,在 18-22 个核苷酸为佳。 退火温度:使多个引物的退火温度尽量保持一致,3-5 ℃ 的差距是可以被接受的。 特异性:引物需要与目标片段互补,并且不与其他区域结合,特异性越高越好。 避免引物二聚体的形成:由于同一个反应体系中存在多个引物,检查引物之间是否会相互配对是十分重要的。 3.9.2 多重 PCR 的优势 内标 internal controls:多重 PCR 由于同时扩增多个目标片段,能够避免普通 PCR 中假阴性的出现。 效率:与使用多管单重 PCR 的系统相比,多重 PCR 的试剂和制备时间的花费更少。当所用 DNA 聚合酶昂贵,或模板稀缺时,多重 PCR 是一个好的选择。 能够指示模板的质量和数量 3.9.3 Relay-PCRRelay-PCR是一种新的、简单且稳健的多重 PCR 方法。一次 PCR 运行完成两个功能独立的反应阶段,无需任何引物去除步骤,从而显着简化了一步工作流程。将一对普通引物和多对特异性引物的混合物添加到单个试管中的基因组 DNA 样品中。捕获感兴趣的基因组靶标的选择阶段和文库扩增阶段都将在这个单一的 PCR 反应期间发生。 更重要的是,通过将特定引物的作用限制在仅用于目标复制的前两个热循环中,并允许在剩余的热循环中自动切换到用于文库扩增的通用引物。这种安排消除了由于特定引物之间的引发效率变化而导致最终产物中扩增子与扩增子变化的重要原因,如果用作扩增引物,这些引物将呈指数级扩增。
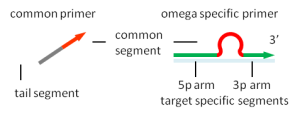
Relay-PCR 使用的 omega 引物:
omega 引物由 3 个功能部分组成: 3p 臂在聚合酶延伸反应中用作引物。 5p 臂用作连接到 DNA 模板的锚。 一个环将两个臂分开。在 Relay-PCR 中,loop 作单重 PCR 的启动部分。
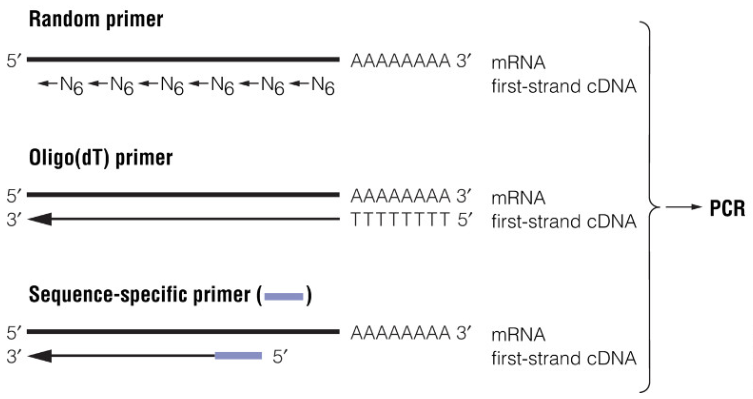
用于基本 PCR 的热稳定性 DNA 聚合酶需要 DNA 模板,因此,该技术仅限于 DNA 样本的分析。要将 PCR 应用于 RNA 研究,必须首先将 RNA 样品转化为 cDNA,以便为热稳定聚合酶提供必要的 DNA 模板。该过程称为逆转录 (reverse transcription, RT),因此此方法被称为 RT-PCR。 3.10.1 RT-PCR 的引物进行逆转录的引物有 3 种类型,取决于实验目的: Oligo(dT) 引物:能结合哺乳动物 mRNA 的 poly(A) 尾,是传统 cDNA 第一链合成的通用引物。 序列特异性引物:cDNA 合成可以由特定的反义寡核苷酸引物引发,该寡核苷酸与特定目标 RNA 或 mRNA 家族中的特定区域特异性杂交。在可能的情况下,应将与位于靶 RNA 不同外显子中的序列结合的寡核苷酸用作扩增 cDNA 产物的正义和反义引物。通过这种方式,可以轻松区分来自 cDNA 和污染基因组 DNA 的扩增产物。然而,无内含子基因的转录本不能与污染基因组序列明确区分开来。在这些情况下,用不含 RNase 的 DNase 处理 RNA 制备可能会有所帮助。 随机的核苷酸六聚体引物:它们能够在沿 RNA 模板的许多点启动 cDNA 合成,生成整个 RNA 分子群的片段拷贝。当目标 RNA 非常长或包含太多二级结构以致 oligo(dT) 或合成寡核苷酸无法有效引发 cDNA 合成时,它们很有用。
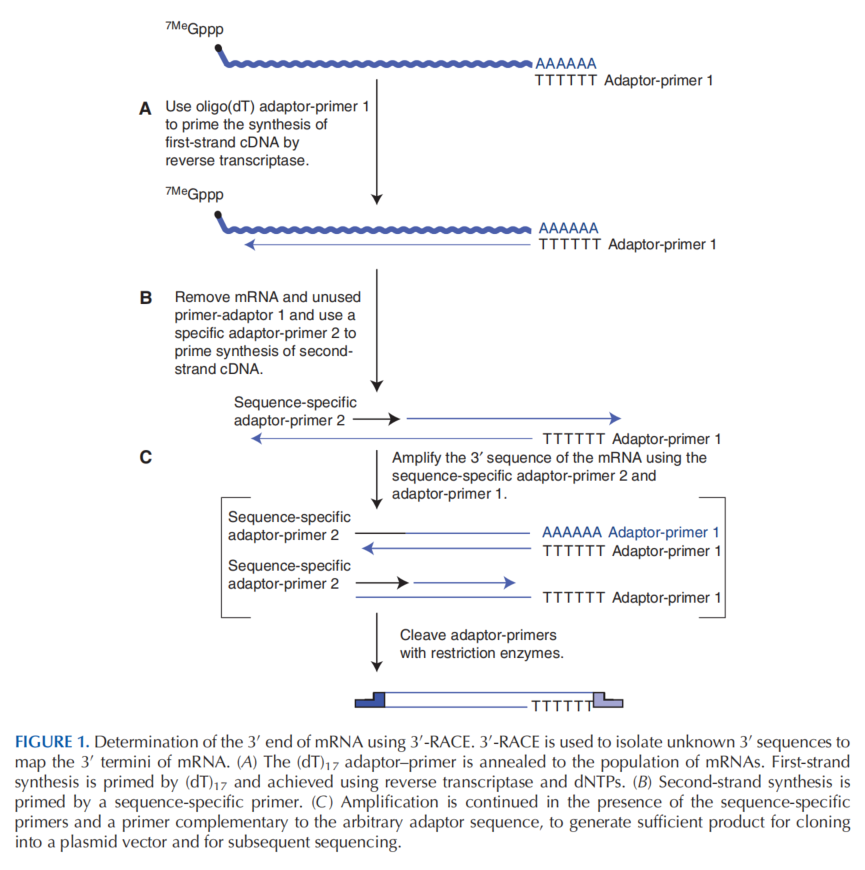
反转录酶(reverse transcriptase, RT):是 RNA 介导的 DNA 合成酶,最初从反转录病毒中分离。RTs 使用反转录引物、dNTPs 和 \(Mg^{2+}\) 或 \(Mn^{2+}\) 作为辅因子催化目标 RNA 分子的 DNA 拷贝 (cDNA) 的合成。 常用的逆转录酶有 3 种: 禽成髓细胞病毒 (AMV),最适温度 42 ℃. 莫洛尼鼠白血病病毒 (M-MLV 或 MuLV),最适温度 37 ℃. 热稳定 rTth DNA 聚合酶,最适温度 60 ℃. 3.10.2.1 AMV 和 MuLV它们可以使用带有 3'-OH 的 RNA 或 DNA 为引物,RNA 或 DNA 为模板,合成 DNA。由于缺乏 3'->5' 核酸外切酶活性,两种酶都是易错的。两种酶催化反应的 Km 值均较高,为了确保 RNA 的完全逆转录,需要使用比基本 PCR 反应更高浓度的 dNTPs. AMV 具有强大的 RNase H 活性,可以消化 RNA-DNA 杂合体的 RNA 部分,如果逆转录酶在合成过程中暂停,它可以在正在生长的 DNA 链 3' 末端附近切割模板。因此,与禽类逆转录酶相关的高水平 RNase H 活性倾向于抑制 cDNA 的产量并限制其长度。 MuLV 逆转录酶比禽类酶更适合 RT-PCR,因为它的 RNase H 活性相对较弱。然而,由于其最适温度为 37 ℃,在此温度下的 RNA 模板二级结构较 42 ℃ 下更多,所以其很容易被 RNA 模板中的二级结构阻断。 许多公司开发了 MuLV 逆转录酶的突变体。与野生型酶相比,修饰的逆转录酶转录更大比例的模板分子并合成更长的 cDNA 分子。此外,它们具有更高热稳定性(高达 60˚C),这在模板 RNA 被折叠成二级结构时是一个优势。这些被改造过的酶被广泛用于一步和两步 RT-PCR。 3.10.2.2 rTth DNA 聚合酶由嗜热细菌 Thermus thermophilus 编码,在 Mn2+ 存在下显示逆转录酶活性。在 RT-PCR 中使用 rTth 聚合酶的主要优点是反应的两个阶段(逆转录和扩增)都在同一个反应管中进行(即一步 RT-PCR)。然而,Tth DNA 聚合酶不能与 oligo(dT) 或随机六聚体一起用作引物,因为杂合体在进行逆转录的温度(60°C 和 70°C 之间)下不稳定。一些制造商出售改进的热稳定 DNA 聚合酶版本,能够有效地将 RNA 复制到 cDNA 中。这些酶主要用于一步法 RT-PCR。 3.10.3 RT-PCR 步骤 释放样品中的 RNA,并进行提纯。 将 RNA 加热以破坏二级结构。 逆转录 RNA 得到 cDNA 的第一条链 将逆转录酶与引物、RNA 模板混合物放在合适的温度进行孵育。 用 95 ℃ 的高温使逆转录酶失活并解开 DNA-RNA 双链。 通过基本 PCR 扩增 cDNA 分析 PCR 产物 3.10.4 One-step & Two-step RT-PCRRT-PCR 可以作为两个独立的反应进行,也可以作为一个需要更特异的商业酶的反应进行。一步法和两步法 RT-PCR 的区别在于:两步法是先完成反转录,反转录引物为通用引物,后以 cDNA 为模板进行后续 PCR 检测;一步法是直接以 RNA 为模板,在一个反应体系中先完成反转录,反转录引物为基因特异性引物(GSP),然后直接进行扩增,得不到cDNA。 两步法 RT-PCR 反转录出的第一链 cDNA 可用作多种扩增反应的底物。 反转录酶一般为最适温度较低,容易被 RNA 中的二级结构阻断反应。一步法 RT-PCR 耦合转录和扩增反应都可以由在高温下具有高活性的热稳定性酶催化。 第一链 cDNA 合成的引发更具特异性,偶联转录反应的创建和优化更快、更简单。 必须为每个反应设置序列特异性引物。 3.10.5 RT-PCR 中的对照 阴性对照 1:包括第一链反应的所有组分,RNA 模板除外。 阴性对照 2:包括第一链反应的所有组分,逆转录酶除外。 阴性对照 3:包括第一链反应的所有组分,引物除外。 阳性对照(可选):包含不同数量的外源参考 RNA,配备一组与真实靶 RNA 中使用的相同的引物退火位点 3.10.6 Ung/dUT 系统Ung/dUT 系统防止非特异性 PCR 扩增和污染。 ung 和 dut 都是大肠杆菌中的基因。UNG 是大肠杆菌的一种尿嘧啶-DNA 糖基化酶 Uracil-DNA glycosylases (UDGs),掺入 DNA 的尿嘧啶残基通常被这种酶去除,该酶切割碱基和脱氧核糖磷酸骨架之间的 N-糖苷键,产生无嘧啶位点。dUT 是脱氧尿苷三磷酸酶 deoxyuridine triphosphatase (dut),一种含锌四聚焦磷酸酶,dUT 能将 dUTP 转化成 dUMP。 UNG 可以特异性降解已经通过 PCR 过程的产物。 UNG 允许先前的 PCR 扩增或错误引发的非特异性产物降解,使用于扩增的天然核酸模板保持完整。 UNG 激活是 PCR 的第一步,在 50°C 孵育 2 分钟。 dUTP:dTTP 比率升高导致 dUTP 在通常由胸腺嘧啶占据的位点的随机子集中错误掺入的频率增加。加入 dUT 能遏制这种现象。 3.11 RACERACE (rapid amplification of cDNA end),已知部分cDNA序列,基于 mRNA 逆转录和 PCR 技术获得完整的 cDNA 的 3' 或5' 末端的分子生物学技术。 3.11.1 3' RACE当采用 PCR 技术进行逆转录时,由寡核苷酸(dT)做引物的第一链 cDNA 的合成通常包含 RNA 的 3' 序列。但是,由序列内部特异性引物做引物合成的 cDNA 通常缺乏 mRNA 两末端的序列,且当 mRNA 含有可变剪接或内部富含 A 时,能够产生不包含 3' 的 cDNA 序列。 含有这些特征的序列能被 3′ -RACE (rapid amplifification of cDNA ends) 技术所恢复,同时,3'-RACE 技术也能被用来将具有可变多聚腺苷酸化位点的 mRNA 家族的 3' 末端比对到基因图上。 3'-RACE 的引物如下: (dT)17 adaptor–primer:其 3' 端具有 poly(T),其 5' 端包含两个或三个限制性内切酶的识别位点。图中为 Adaptor-primer 1。 序列特异性引物 sequence-specific primer(基因特异性引物 gene-specific primer, GSP):与 mRNA 内基因互补,可带有限制性内切酶位点。图中为 Adaptor-primer 2.采用 (dT)17 adaptor–primer 将一组 mRNA 逆转录成 cDNA。逆转录后通过使用序列特异性引物转变为双链 DNA。扩增的产物被分离、用合适的限制酶切割、克隆和表征。 在一轮 PCR 结束后,还可以使用嵌套 PCR,以提高 cDNA 的丰度。
在 cDNA 建库中,缺少 5' mRNA 序列的 cDNA 序列经常出现。5'-RACE 与 3'-RACE 类似,其能在已知少部分序列的情况下,扩增 mRNA 5' 端序列。其根据已知的 cDNA 序列设计基因特异性引物,在逆转录酶的作用下,合成第一条链 cDNA,末端转移酶在 cDNA 链 3' 端加入连续的同聚核苷酸,以连有互补配对的同聚核苷酸引物和基因特异性引物进行 PCR 扩增,以期得到 mRNA 的 5' 端。 5'-RACE 流程: 逆转录 同聚物加尾:采用末端转移酶 PCR 扩增5'-RACE 的引物如下: 序列特异性引物 sequence-specific primer(基因特异性引物 gene-specific primer, GSP):与 mRNA 内基因互补,图中为 Sequence-specific primer 1. 可带有限制性内切酶位点,图中为 Adaptor-primer 2. (dT)17 adaptor–primer:其 3' 端具有 poly(T),其 5' 端包含两个或三个限制性内切酶的识别位点。图中为 Adaptor-primer 1.
末端转移酶:末端脱氧核糖核苷酸转移酶 (Tdt),一种 510 个氨基酸的单体酶,能在没有模板的情况下,催化数百个 dNTPs 添加到 DNA 分子的 3' 末端。在分子克隆中,末端转移酶主要用于催化在 5'-RACE 和 3'-RACE 反应中产生的单链 DNA 中添加同聚尾。当面对不同的具有 3' 末端的 DNA 分子时,末端转移酶倾向于对带有未配对的 3'-DNA 加尾,其次是平末端 DNA,最后是凹陷的 3' 末端。在平末端的情况下,添加第一个核苷酸是反应的限速步骤。 末端转移酶对反应条件有不寻常的要求。它被反应缓冲液中通常包含的许多阳离子所抑制,包括铵离子、氯离子和磷酸根离子。大多数反应在甲次砷酸盐(即二甲基砷酸)缓冲液中进行,100 mM Tris-乙酸盐(pH 7.2)也可。使用 dTTP 或 dCTP 作为底物的均聚拖尾反应在 \(Co^{2+}\) 存在下进行; \(Mn^{2+}\) 是涉及嘌呤残基(A/T)聚合的均聚拖尾反应的首选辅助因子。 末端转移酶在 pre-B 和 pre-T 淋巴细胞中表达,通常用作由这些细胞引起的某些类型白血病的标志物。在免疫系统成熟期间,Tdt 通过添加不依赖模板的核苷酸和破坏同源定向重组来介导 T 细胞受体库的扩增和多样化 3.12 Real-time Quantitative PCR (real-time qPCR)检测目标 RNA 高准确度和敏感度的方法,即使目标 RNA 在细胞中微量存在。real-time qPCR 是所有检测 RNA 方法中最敏感、快速、准确的,在检测不同细胞或组织中的 mRNA 相对丰度时最主流的方法。Real-time qPCR 不仅仅能够检测 mRNA,其也能检测 microRNA 丰度。 不同的商用产品采用不同的检测方法,有上百种检测的流程。real-time qPCR 每步缺乏标准化,使结果的可信度和可重复性下降。在发表结果时,需要附带一系列的必选实验参数,以增加结果的可信度和可重复性。 3.12.1 qRT-PCR 的步骤 制备高质量的 RNA。保证纯度和完整性。 使用依赖于 RNA 的 DNA 聚合酶(逆转录酶)将 RNA 转化为互补 DNA (cDNA)。 引发方法按照流行度排序:oligo(dT), random priming, gene-specific priming. cDNA 通过热稳定的DNA聚合酶扩增。 3.12.2 qRT-PCR 的内参内参选择原则: 在测定的所有样品中表达量恒定。 不受实验中变量的影响。内参举例: β-actin:在不同的白血病肿瘤样本中差异表达。 glyceraldehyde-3-phosphate dehydrogenase (GAPDH):在某些侵袭性癌症中升高。 hypoxanthine-guanine phosphoribosyl transferase (HPRT):在大多数人体组织中以低水平组成性表达,但在中枢神经系统的某些部分中以高水平表达。 18S ribosomal RNA:在巨细胞病毒感染时表达量上升。 Polymerase Chain Reaction (PCR) Amplification of cDNA Generated by Reverse Transcription of mRNA: Two-Step Reverse Transcription-Polymerase Chain Reaction (RT-PCR) Quantification of RNA by Real-Time Reverse Transcription-Polymerase Chain Reaction (RT-PCR) Rapid Amplification of Sequences from the 5′ Ends of mRNAs: 5′-RACE Rapid Amplification of Sequences from the 3′ Ends of mRNAs: 3′-RACE Analysis and Normalization of Real-Time Polymerase Chain Reaction (PCR) Experimental Data Enzymes used in PCR (qiagen.com) PCR Troubleshooting (caister.com) DNA聚合酶—关于PCR的四大关键属性 | Thermo Fisher Scientific - CN PCR反应中各种添加剂的作用 - 知乎 (zhihu.com) PCR Amplification | An Introduction to PCR Methods | Promega Stimulatory effect of potassium glutamate in PCR - PubMed (nih.gov) Replacement of potassium chloride by potassium glutamate dramatically enhances protein-DNA interactions in vitro - PubMed (nih.gov) Cleavage Close to the End of DNA Fragments | NEB Relay-PCR™ - LC Sciences - Technologies for Genomics & Proteomics Discoveries |

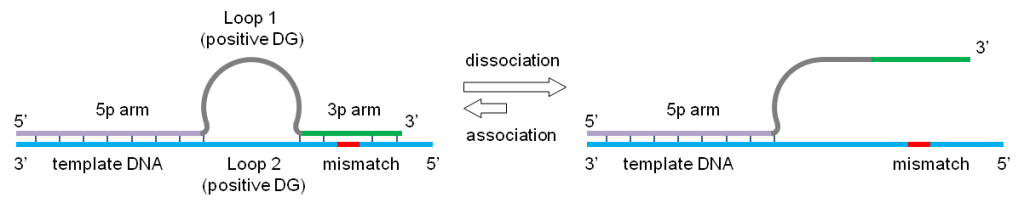

【本文地址】