| 【精选】做折线图位置引用无效 | 您所在的位置:网站首页 › excel表格折线图位置引用无效 › 【精选】做折线图位置引用无效 |
【精选】做折线图位置引用无效
|
紧接着之前的文章【如何去除核糖体RNA(rRNA)序列?】中,使用比对到rRNA上的bam文件,进行样本rRNA深度折线图的绘制。 Kevin.Lau:【生信】【转录组】如何去除核糖体RNA(rRNA)序列?zhuanlan.zhihu.com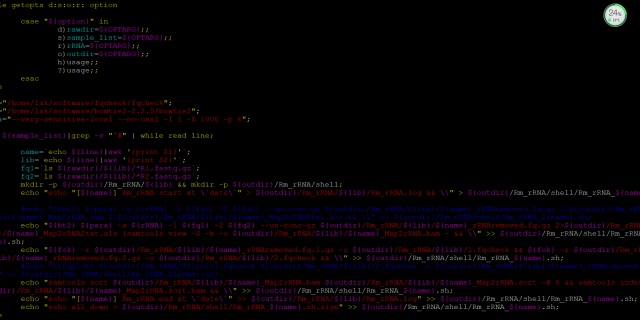
在文章中,bowtie2比对后,同时得到了: *rRNAremoved.fq.1.gz和*rRNAremoved.fq.2.gz*Map2rRNA.bam其中2就是样本中核糖体RNA序列的bam文件,我们可以看看样本中不同的rRNA的深度如何分布,所谓“一图胜前言”,于是,尝试绘制其深度折线图。 首先,在【如何去除核糖体RNA(rRNA)序列?】一文中,提取了rRNA的参考序列之后,如果要做这个折线图,建议直接在参考序列fa文件中修改各个rRNA的id信息行,再进行bowtie2去除rRNA,便于后续的制图。 rRNA原始id,没办法直接看出对应的序列是哪种rRNA,因为bowtie2中会将id信息体现在bam中,对原始id进行修改: cat GCF_000001405.25_GRCh37.p13_rna_from_genomic.fna | grep "^>" | grep "gbkey=rRNA" | awk '{print $1}'|less -S >lcl|NC_000001.10_rrna_NR_023363.1_7456 >lcl|NC_000001.10_rrna_NR_023364.1_7457 >lcl|NC_000001.10_rrna_NR_023365.1_7458 >lcl|NC_000001.10_rrna_NR_023366.1_7459 >lcl|NC_000001.10_rrna_NR_023367.1_7460 >lcl|NC_000001.10_rrna_NR_023368.1_7461 >lcl|NC_000001.10_rrna_NR_023369.1_7462 >lcl|NC_000001.10_rrna_NR_023370.1_7463 >lcl|NC_000001.10_rrna_NR_023371.1_7464 >lcl|NC_000001.10_rrna_NR_023372.1_7465 >lcl|NC_000001.10_rrna_NR_023373.1_7466 >lcl|NC_000001.10_rrna_NR_023374.1_7467 >lcl|NC_000001.10_rrna_NR_023375.1_7468 >lcl|NC_000001.10_rrna_NR_023376.1_7469 >lcl|NC_000001.10_rrna_NR_023377.1_7470 >lcl|NC_000001.10_rrna_NR_023378.1_7471 >lcl|NC_000001.10_rrna_NR_023379.1_7474 >lcl|NT_167214.1_rrna_NR_046235.3_81980 >lcl|NT_167214.1_rrna_NR_003286.4_81981 >lcl|NT_167214.1_rrna_NR_003285.3_81982 >lcl|NT_167214.1_rrna_NR_003287.4_81983 >lcl|NC_012920.1_rrna_90943 >lcl|NC_012920.1_rrna_90945前文提到5S rRNA的17条序列一模一样,去冗余,根据信息行,修改成如下id,加上对应的类型: cat rRNA.fa | grep "^>" | awk '{print $1}'|less -S >NR_023363.1_5S >NR_046235.3_45S >NR_003286.4_18S >NR_003285.3_5.8S >NR_003287.4_28S >MT.rrna_12S >MT.rrna_16Sbowtie2得到Map2rRNA.bam后,使用samtools sort 进行排序,使用samtools index构建索引。 samtools sort Map2rRNA.bam Map2rRNA.sort -@ 4 && samtools index Map2rRNA.sort.bam排序并且构建索引后,使用samtools depth对Map2rRNA.sort.bam文件计算各rRNA的深度信息,下面是samtools depth的使用方法: Usage: samtools depth [options] in1.bam [in2.bam [...]] Options: -b list of positions or regions -f list of input BAM filenames, one per line [null] -l minQLen -q base quality threshold -Q mapping quality threshold -r region其中-b是我们需要计算深度的区域的bed文件,在本文中,因为我们要看rRNA全长的深度,所以只需要得到每个rRNA的长度L,就可以做出rRNA.bed文件。需要注意的是,samtools depth中的bed文件是0-base的。 写个python小脚本来得到rRNA.bed文件: # -*- coding:UTF-8 -*- import sys def main(fq): with open(fq,'r') as f: f_tmp = f.readlines() f_dict = {} for i in f_tmp: if i.startswith('>'): trid = i.split()[0][1:] f_dict[trid] = 0 else: f_dict[trid] += len(i.strip()) for k,v in f_dict.items(): print k + 't'+ '0' + 't' + str(v) #也可以只得到rRNA的长度信息 #print k + 't' + str(v) if __name__ == "__main__": fq = sys.argv[1] main(fq)运行python小脚本: python2 fa_idlen.py rRNA.fa > rRNA.bed cat rRNA.bed NR_023363.1_5S 0 121 NR_003285.3_5.8S 0 157 MT.rrna_16S 0 1559 NR_046235.3_45S 0 13357 NR_003287.4_28S 0 5070 NR_003286.4_18S 0 1869 MT.rrna_12S 0 954顺便可以把rRNA的长度信息做成一个文件rRNA.len,其内容就是rRNA.bed的第1、3列。 samtools depth计算rRNA深度: samtools depth -b rRNA.bed Map2rRNA.sort.bam > Map2rRNA.sort.depth.txt head Map2rRNA.sort.depth.txtMap2rRNA.sort.depth.txt的第一列是rRNAid,第二列是位置,第三列是该位置的碱基数(即深度),部分结果如下: NR_023363.1_5S 1 10 NR_023363.1_5S 2 11 NR_023363.1_5S 3 16 NR_023363.1_5S 4 20 NR_023363.1_5S 5 49 NR_023363.1_5S 6 64 NR_023363.1_5S 7 142 NR_023363.1_5S 8 1302 NR_023363.1_5S 9 1749 NR_023363.1_5S 10 2026此时,样本rRNA的深度信息已经得到,接下来就是绘制其深度的折线图。 样本的深度信息文件中,有时候并不包含所有的rRNA位点,当某区域深度为0时,这部分区域是不会输出到深度文件中的。比如,如果 NR_023363.1_5S:0-7 这个位置没有任何reads落在上面,那么前面的部分结果就会显示为: NR_023363.1_5S 8 1302 NR_023363.1_5S 9 1749 NR_023363.1_5S 10 2026前面的7碱基不会输出为0,而是直接不输出内容,因此在绘图时需要注意,要补上这些深度为0的区域。另外,绘图中,X轴的位置如果都按照实际的位置去画的话,那么各个rRNA的折线就会在X轴上重叠,可以考虑适当处理一下X轴的位置坐标,使得每个rRNA的折线不重叠。 经过X轴位置处理的depth.txt文件用于后续绘图。 使用R包,ggplot2进行折线图绘制: #载入R包ggplot2 library(ggplot2) #载入深度信息文件depth.txt,添加列名 rRNA |
【本文地址】