| GSEA确实搭配热图后更直观易懂 | 您所在的位置:网站首页 › es和ls的区别 › GSEA确实搭配热图后更直观易懂 |
GSEA确实搭配热图后更直观易懂
|
最近学徒被委托模仿一个2022的发表在Biomaterials杂志的文章《Engineering dual catalytic nanomedicine for autophagy-augmented and ferroptosis-involved cancer nanotherapy》的转录组两分组标准分析以及出图,我们默认流程是差异分析和生物学功能数据库注释的富集分析。 其中生物学功能数据库注释目前最稳妥的是GSEA方法,但是文章在标准的gsea图下面加上了一个热图,蛮有意思的:  gsea图下面加上了一个热图 gsea图下面加上了一个热图因为纯粹的GSEA方法出图后很多人都没办法理解。 假设芯片或者其它测量方法测到了2万个基因,那么这两万个基因在case和control组的差异度量 (其实有六种差异度量,默认是signal 2 noise,GSEA官网有提供公式,也可以选择大家熟悉的foldchange) 肯定不一样, 那么根据它们的差异度量,就可以对它们进行排序,并且Z-score标准化,在GSEA图的最底端展示的就是排序后的基因列表 : 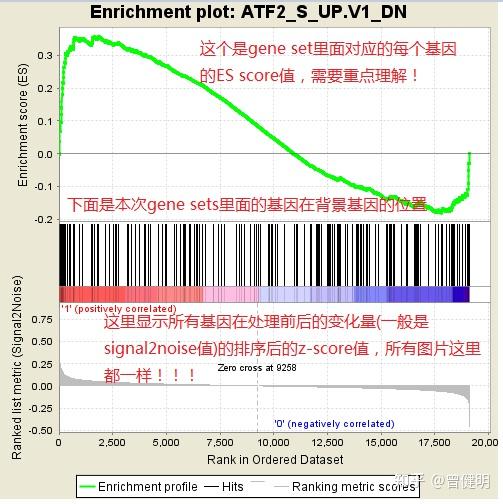 纯粹的GSEA方法出图 纯粹的GSEA方法出图那么图中间,就是我们每个gene set里面的基因(通常是几十个基因甚至上百个基因)在所有的2万个排序好基因的位置,如果gene set里面的基因集中在2万个基因的前面部分,就是在case里面富集,如果集中在后面部分,就是在control里面富集着。 而最上面的那个ES score的算法,大概如下:  ES score的算法 ES score的算法仔细看,其实还是能看明白的,排序好的2万多个基因里面的每个基因在每次gsea分析的每个gene set里面的ES score取决于这个基因是否属于该gene set,还有就是它的差异度量,上图的差异度量就是FC(foldchange), 对每个gene set来说,所有的基因的ES score都要一个个加起来,叫做running ES score,在加的过程中,什么时候ES score达到了最大值,就是这个gene set最终的ES score ! 所以实际上大家只需要看每个gene set最终的ES score即可,比如: library(clusterProfiler) library(org.Hs.eg.db) library(DOSE) data(geneList) head(geneList) #排序好的基因序列,而且是entrezeID的形式 R.utils::setOption( "clusterProfiler.download.method",'auto' ) kkgsea image-20221030153240324我们常规的可视化代码就是enrichplot包的gseaplot2函数,代码如下所示: library(enrichplot) gseaplot2(kkgsea,geneSetID = head(kkgsea@result$ID,12))显示12个通路,这样可以看到正负值的区别, 可视化效果; 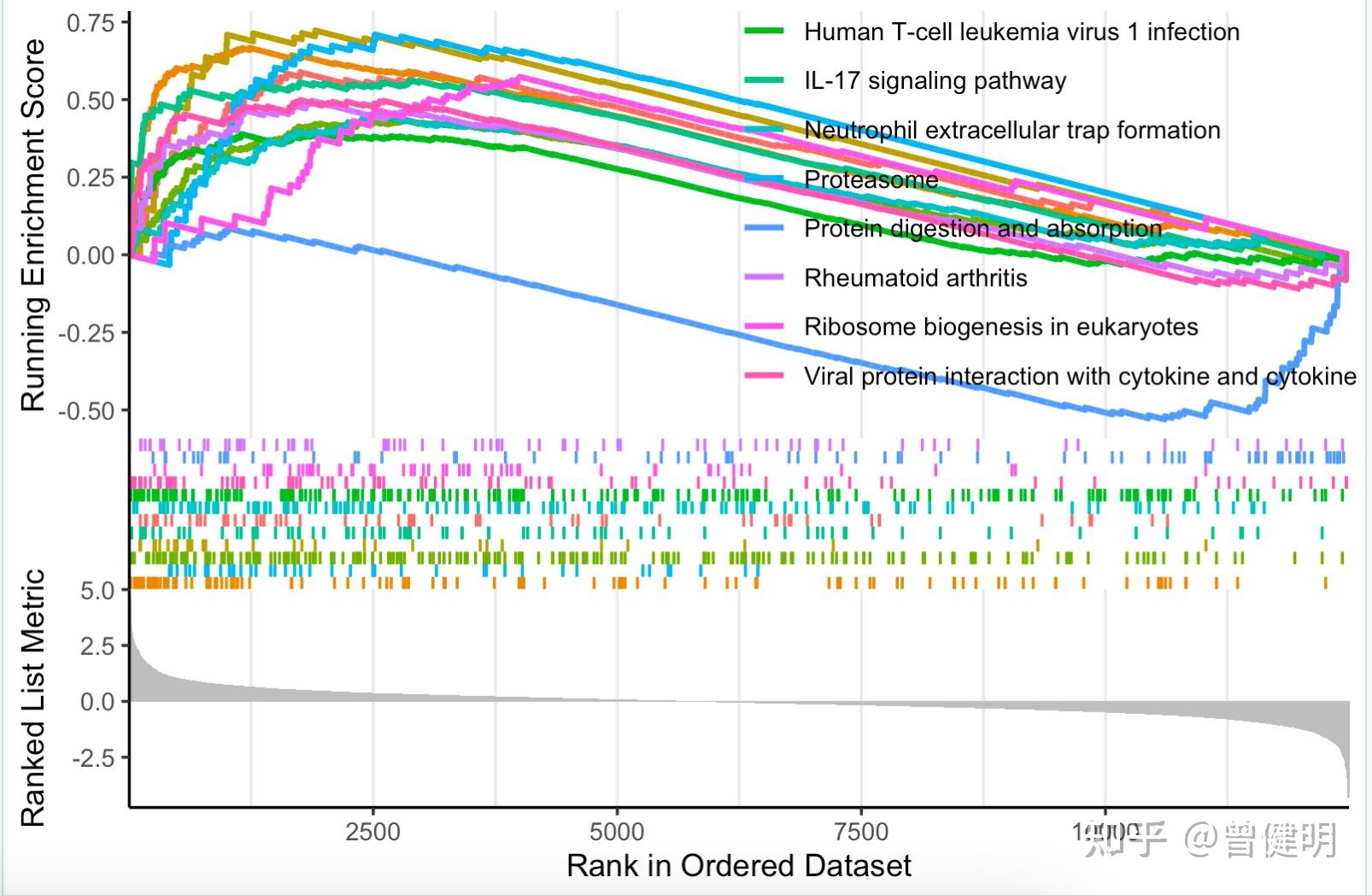 看到正负值的区别 看到正负值的区别上面的gsea和分析和可视化是直接使用了DOSE里面的data(geneList) ,这个排序好的基因序列,而且是entrezeID的形式。 实际上我们自己的 基因排序,往往是来自于自己的差异分析后的: rm(list = ls()) options(stringsAsFactors = F) load(file = "./data/Step03-DESeq2_nrDEG.Rdata") load(file = "./data/Step01-airwayData.Rdata") head(deg_anno) library(clusterProfiler) library(org.Hs.eg.db) # 转成ENTREZID df |
【本文地址】
| 今日新闻 |
| 推荐新闻 |
| 专题文章 |