| picrust2功能预测 | 您所在的位置:网站首页 › biom安装 › picrust2功能预测 |
picrust2功能预测
|
记录一下跌跌撞撞地摸索过程 picrust2功能预测 参照公众号《宏基因组》刘永鑫的原创:《PICRUSt2:OTU或ASV等16S随便预测宏基因组且数据库增加10倍》https://blog.csdn.net/woodcorpse/article/details/89302863 注:命令中 \为断行用,实际上输入命令要去掉,在输入文件路径时没有用此符号断行。 虚拟机与qiime2安装 参照公众号《宏基因组》刘永鑫的原创:《QIIME 2用户文档. 01简介和安装 Introduction & Install(2020.2) 》使用VirtualBox方式安装部分 https://blog.csdn.net/woodcorpse/article/details/75103929 下载最新版本虚拟机:https://download.virtualbox.org/virtualbox/6.1.4/VirtualBox-6.1.4-136177-Win.exe 下载最新版本虚拟机扩展包: https://download.virtualbox.org/virtualbox/6.1.4/Oracle_VM_VirtualBox_Extension_Pack-6.1.4.vbox-extpack 下载QIIME2镜像:https://data.qiime2.org/distro/core/2020.2 安装: 1) 安装最新版虚拟机,默认位置,装好后将扩展包拷贝到安装目录windows/program files/oracle/virtualbox,双击安装扩展包。 2) 双击下载好的QIIME2镜像或在虚拟机界面点击导入镜像,修改分配给虚拟机的cpu和内存(一般是windows的一半),点击导入。 3) 启动虚拟机,菜单中安装Guest Additions,按照提示完成后,重启linux系统才能设置共享文件夹和共享复制粘贴。https://mp.weixin.qq.com/s/WS9u0nhiS1eizL5KXKs__A? 4) 设置共享文件夹,添加共享文件夹,勾选固定分配/自动挂载。 问题1:安装好后,输入qiime的命令均显示command not found。 前几天看到有人在百度上发起了同样问题。不是安装有问题,是qiime的命令和qiime2的命令不兼容,因此使用qiime2进行16分析的命令可参照https://www.cnblogs.com/afeiyuanda/p/11037287.html Picrust2安装 https://github.com/picrust/picrust2/官网有给出两种安装方法,建议第一种安装,不会出错。 安装好后第二次使用picrust,也要输入激活命令: conda activate picrust2 或者 conda activate picrust如果想要使用qiime2命令,则要先退出picrust2,输入失活命令: conda deactivate问题2:v2.3.2-b版本优化了匹配度,测序序列进行后续分析时都会出现这样的问题,stopping - all 234 input sequences aligned poorly to reference sequence(--min_align option specified a minimum proportion of 0.8 aligning to reference sequences 先反向互补完,再做功能预测就可以了,seqtk seq -r otus.fa.format.fasta > otus_rc.fa
如果使用以前的版本不会出现这样的问题。 有个小插曲:不知道怎么解决这个问题的时候,无论输入什么数据都显示出这个结果,心态差点就崩了,然后花了点钱请淘宝上做生信分析的人,用我的数据帮我跑了一遍,可以出结果。最后观察版本之间有什么差别的时候就发现最新版本优化了,然后淘宝上那个生信技术厉害的客服用最新版本也出不了结果,他就去官网发了一个issue(就是上面图片里的),开发者很快就给回复了,解释了原因。 问题3:使用vsearch跑出的结果使用out.fa和out_table.biom两个文件可以直接跑出预测结果,但使用qiime跑出的结果中两个文件夹的out名称不一致,要删掉后面的其他字符可采用以下方法。 修改fastq文件OTU 名字` awk '{print $1 }' FASTA_IN > FASTA_OUT其中FASTA_IN和FASTA_OUT是输入和输出FASTA文件的名称。 $2:一行一行的读取指定的文件, 以空格作为分隔符,打印第二个字段 比如有这样一个文件 a1 b1 c1 d1 a2 b2 c2 d2 执行的结果是,输出: b1 b2 问题4:会出现以下报错,大概率是电脑内存不够,picrust2官网有一个针对这个错误的issue。
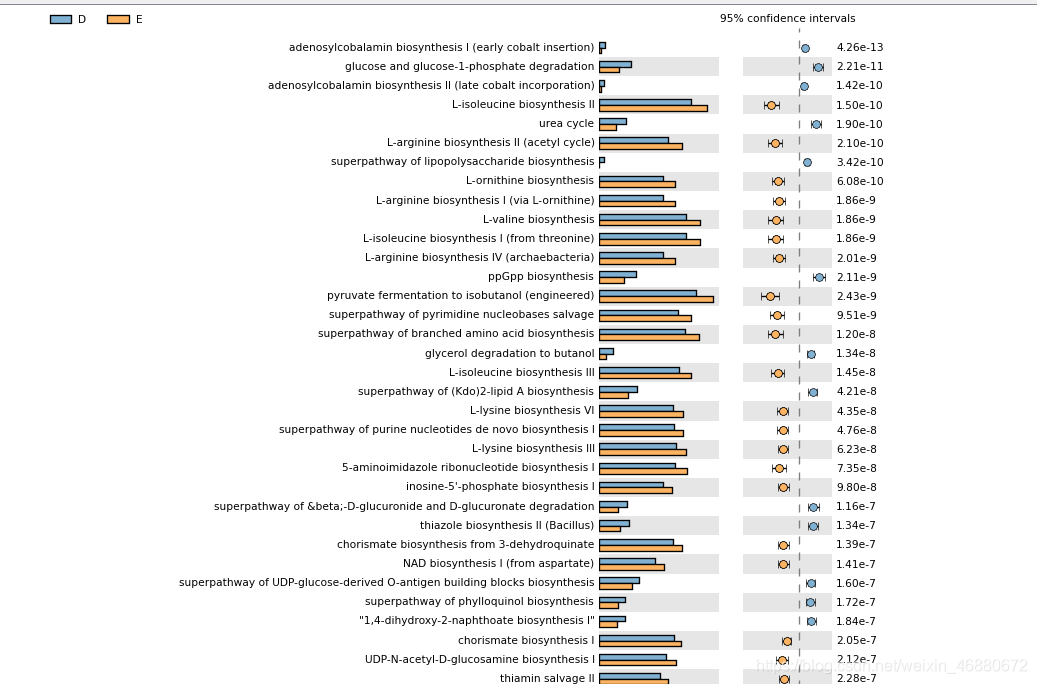
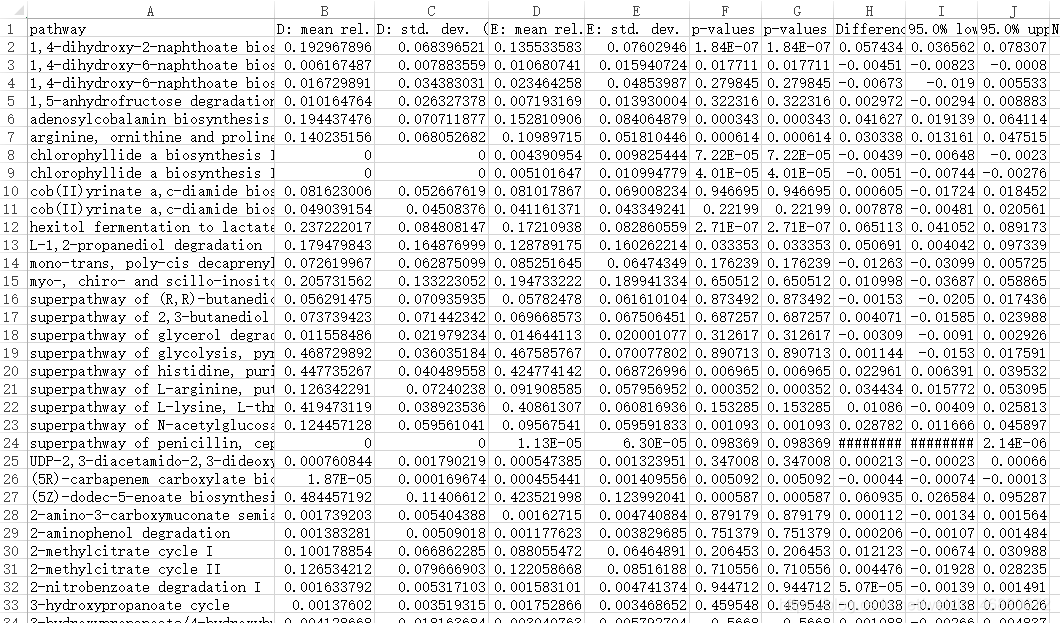
解决的根本办法是扩大你的内存,建议32G往上走,windows32G内存一般能够给虚拟机分配20G左右运行内存,在样本量比较多的情况下,往往16G是不够的,但是有时候样本量可以的情况下可以打擦边球试运行,降低CPU线程。我之前运行过分配给虚拟机36G的内存(windows总共48G)运行104个样本,使用4和2线程均无法得到结果,之后再使用1个线程耗时19000秒就成功了。也有试过88个样本,2线程9000秒。 ###picrust2预测 比较完整的一步脚本是 picrust2_pipeline.py -s dna-sequences.fasta -i feature-table.biom -o \ picrust2_out_pipeline -p 20 -r default_files/prokaryotic/pro_ref/pro_ref \ --in_traits COG,EC,KO,PFAM,TIGRFAM可能default_files/prokaryotic/pro_ref/pro_ref这个文件需要你在你的虚拟机上搜索后输入完整并且正确的文件路径。 这个命令运行后的结果包括COG,EC,KO,PFAM,TIGRFAM的丰度信息和MetaCyc途径丰度。 我一般输入的命令是: picrust2_pipeline.py –s out.fa -i out_table.biom \ -o picrust2_out_pipeline -p 4 -p 4是使用4线程的意思输出的结果包括EC,KO,PATHWAY的丰度信息。 但是但是结果文件中的结果比较简洁,比如只输入EC1.1.1,而不标注对应的酶名字,这时候需要加工加工。根据需要酌情添加描述: add_descriptions.py -i EC_metagenome_out/pred_metagenome_unstrat.tsv \ -m EC -o EC_metagenome_out/pred_metagenome_unstrat_descrip.tsv add_descriptions.py -i KO_metagenome_out/pred_metagenome_unstrat.tsv \ -m KO -o KO_metagenome_out/pred_metagenome_unstrat_descrip.tsv add_descriptions.py -i COG_metagenome_out/pred_metagenome_unstrat.tsv \ -m COG -o COG_metagenome_out/pred_metagenome_unstrat_descrip.tsv add_descriptions.py -i PFAM_metagenome_out/pred_metagenome_unstrat.tsv \ -m PFAM -o PFAM_metagenome_out/pred_metagenome_unstrat_descrip.tsv add_descriptions.py -i TIGRFAM_metagenome_out/pred_metagenome_unstrat.tsv\ -m TIGRFAM -o TIGRFAM_metagenome_out/pred_metagenome_unstrat_descrip.tsv add_descriptions.py -i pathways_out/path_abun_unstrat.tsv -m METACYC -o pathways_out/path_abun_unstrat_descrip.tsv结果输出中没有KEGG结果,可通过以下命令生成KEGG pathway 丰度表和注释结果 pathway_pipeline.py -i KO_metagenome_out/pred_metagenome_unstrat.tsv \ -o KEGG_pathways_out --no_regroup --map \ /home/qiime2/miniconda/envs/picrust/lib/python3.6/site-package/ picrust2/default_files/pathway_mapfiles/KEGG_pathways_to_KO.tsv 这里KEGG_pathways_to_KO.tsv需要你在你的虚拟机上搜索后输入完整并且正确的文件路径。 # 添加功能描述 add_descriptions.py -i KEGG_pathways_out/path_abun_unstrat.tsv.gz \ --custom_map_table /home/qiime2/miniconda/envs/picrust/lib/python3.6/site- package/picrust2/default_files/description_mapfiles/KEGG_pathways_info.tsv.gz \ -o KEGG_pathways_out/path_abun_unstrat_descrip.tsv.gz 这里/KEGG_pathways_info.tsv.gz需要你在你的虚拟机上搜索后输入完整并且正确的文件路径。如果不懂可看原文https://www.jianshu.com/p/e3beaee77423 或如果不想一步生成结果,就看官网分步命令:https://github.com/picrust/picrust2/wiki/PICRUSt2-Tutorial-(v2.1.4-beta) 分析好的结果,使用STAMP软件进行显著性差异分析,该软件导入相应数据后既可以直接出很直观漂亮的显著性差异图,也可一次性导出所有通路或酶的组间差异分析详细数据。如下两图所示。 |
【本文地址】